
聚噻吩的后修饰与光电性质
2021-10-22 11:58:02谢子益卫青云钟洪亮
功能高分子学报 2021年5期
谢子益, 单 通, 卫青云, 钟洪亮
(上海交通大学化学化工学院,上海 200240)
有机太阳能电池(OSCs)具有柔性、轻质、可溶液加工、高通量制备等优点,是新一代的绿色能源[1-4]。为了进一步推动其商业化应用,降低OSCs的成本,特别是开发低成本且易于制备的光伏材料至关重要。其中聚噻吩材料(如聚3己基噻吩(P3HT))可以通过Kumada催化剂转移缩聚(Kumada catalyst transfer polycondensation, KCTP)反应制备,是目前少数已商业化的光伏材料系列之一[4-6]。KCTP法采用格氏试剂和镍催化剂价格便宜,此外,其聚合机理为链增长,可以得到具有可控分子量和低分散度的共轭聚合物材料,适合于大规模工业化生产[7-9]。但是KTCP法所用的格氏试剂具有较强的化学反应活性,易与缺电子基团发生反应,同时,催化剂转移的机理也限制了单体的共轭长度,使得KTCP法仅适用于共轭长度小的富电子芳香环[10,11],造成聚噻吩的化学结构比较单一,在能级、吸光、电荷传输等方面的局限性较大,且不易被化学修饰,限制了其在光电器件中的应用。
为了改善聚噻吩材料的光电性质,前期研究将缺电子官能团引入到噻吩单体中,通过Stille偶联反应构建了新型聚噻吩材料。缺电子官能团的引入有效促进了聚合物分子内的电荷转移,降低了前线轨道能级,拓宽了吸光范围[12,13]。2009年,侯剑辉课题组[14]在聚噻吩材料中引入了酯基官能团,所构建聚合物相比于P3HT有着更为合适的轨道能级,当与非富勒烯受体分子匹配时,光伏器件效率超过了10%。但是利用Stille偶联反应合成聚合物也存在相应的问题,采用的有机锡试剂毒性较大,钯催化剂价格昂贵,不利于大规模工业生产。
针对以上挑战,利用简捷的KCTP合成方法,引入缺电子基团对聚噻吩进行修饰,可继续发展新的材料设计与合成策略。因此,本文利用KCTP法构建了含有硫代烷基侧链的聚噻吩(PtTSBO),通过控制氧化剂间氯过氧苯甲酸(m-CPBA)的用量对侧链的硫醚进行氧化[15],获得相应的含有亚砜和砜基的聚噻吩材料(PtTSOBO和PtTSOOBO)。分析结果表明,后修饰策略切实可行。进一步的光电性质研究显示,亚砜和砜基的引入改变了聚噻吩的电荷分布,降低了前线轨道能级,从而大幅提升了OSCs器件的开路电压(Voc)。这种后修饰策略对于改性聚噻吩,推动功能化聚噻吩材料的低成本生产提供了新思路。
1 实验部分
1.1 原料和试剂
间氯过氧苯甲酸(m-CPBA):分析纯,上海麦克林生化科技有限公司;N-溴代丁二酰亚胺(NBS)、碳酸钾、2-噻吩硼酸频哪醇酯:分析纯,上海毕得医药科技有限公司;2,2,6,6-四甲基哌啶基氯化镁氯化锂(TMPMgCl·LiCl)、[1,3-双(二苯基膦基)丙烷]二氯化镍(Ni(dppp)Cl2)、四正丁基六氟磷酸铵(n-Bu4NPF6):分析纯,萨恩化学技术有限公司;无水氯化铜:分析纯,北京伊诺凯科技有限公司;甲苯(Toluene)、四氢呋喃(THF)、乙腈、正己烷、盐酸:分析纯,国药集团化学试剂有限公司;氘代氯仿(CDCl3)、氘代四氯乙烷(C2D2Cl4)、四合三苯基膦钯(Pd(PPh3)4):分析纯,上海百灵威化学技术有限公司;石油醚、二氯甲烷、氯仿(CHCl3)、甲醇、氢化钙、无水硫酸钠:分析纯,上海泰坦科技股份有限公司;以上试剂无特殊说明直接使用。用于层析柱分离提纯的硅胶:分析纯,粒度为38~49 μm,青岛海洋化工试剂有限公司;无水四氢呋喃:在氩气氛围下,以THF、金属钠和二苯甲酮为指示剂,回流搅拌直至溶液呈蓝紫色,收集馏分后备用。
1.2 测试与表征
核磁共振氢谱(1H-NMR)和碳谱(13C-NMR):德国布鲁克公司AVANCE III HD 400型核磁共振波谱仪,所用的试剂为氘代氯仿和氘代四氯乙烷,温度为60 ℃。
凝胶渗透色谱(GPC):日本岛津公司GPC-20A型,测试温度为80 ℃,以氯苯为流动相、聚苯乙烯为内标,测试数均分子量(Mn)、重均分子量(Mw)和分子量分布(Đ=Mw/Mn)。
元素分析(EA):美国ThermoFisher公司FlashSmart型元素分析仪。
紫外-可见分光(UV-Vis)光谱:美国珀金埃尔默公司Lambda 750S型紫外-可见分光光度计,聚合物稀溶液的光谱吸收是将样品溶解在氯仿溶液中,配制成7×10-4mg/mL的溶液进行测试;聚合物膜的光谱吸收是将聚合物的氯仿溶液(8 mg/mL)旋涂在载玻片上进行测试。
基质辅助激光解吸电离飞行时间质谱(MALDI-TOF MS):德国布鲁克公司Autoflex speed TOF/TOF型MALDI-TOF MS仪。采用反射模式,将所测单体配制成2 mol/L的二氯甲烷溶液进行点样,基质为反式-2-[3-(4-叔丁基苯基)-2-甲基-2-丙烯基]丙二腈(DCTB)。
电化学循环伏安(CV)曲线:上海振华CHI604E型电化学工作站,采用三电极体系,参比电极为Ag/AgCl电极,对电极为铂片电极,涂有聚合物的氧化铟锡(ITO)玻璃作为工作电极。二茂铁(Fc/Fc+)作为内标,电解质溶液为n-Bu4NPF6的乙腈溶液(c=0.1 mol/L)。
原子力显微镜(AFM):德国布鲁克公司Multimode 8型原子力显微镜,在轻敲模式(Tapping mode)下测得活性层表面形貌的高度图与相图。
密度泛函理论(DFT)计算:在模拟过程中,为了简化计算,将聚合物的烷基侧链简化为甲基,通过Gaussian 09软件在B3LYP/6-31G模型下计算得到最优构象,并计算出理论最高占有轨道(HOMO)和最低未占有轨道(LUMO)电子云分布和能级。
有机太阳能电池测试:通过美国Keithley公司 2 420 Source Meter数字源表测得光伏器件的电流密度-电压(J-V)特性曲线,采用中国光焱科技Enlitech SS-F5-3A型太阳能模拟器模拟太阳光(AM 1.5G),光照强度为100 mW/cm2。
1.3 实验步骤
1.3.1 聚合物PtTSBO的合成 2-溴-3-(硫代(2-丁基-辛基))-噻吩(标记为1)参考文献报道合成[16,17],并通过核磁确证结构。聚合物PtTSBO的合成路线见图1,具体合成步骤如下。
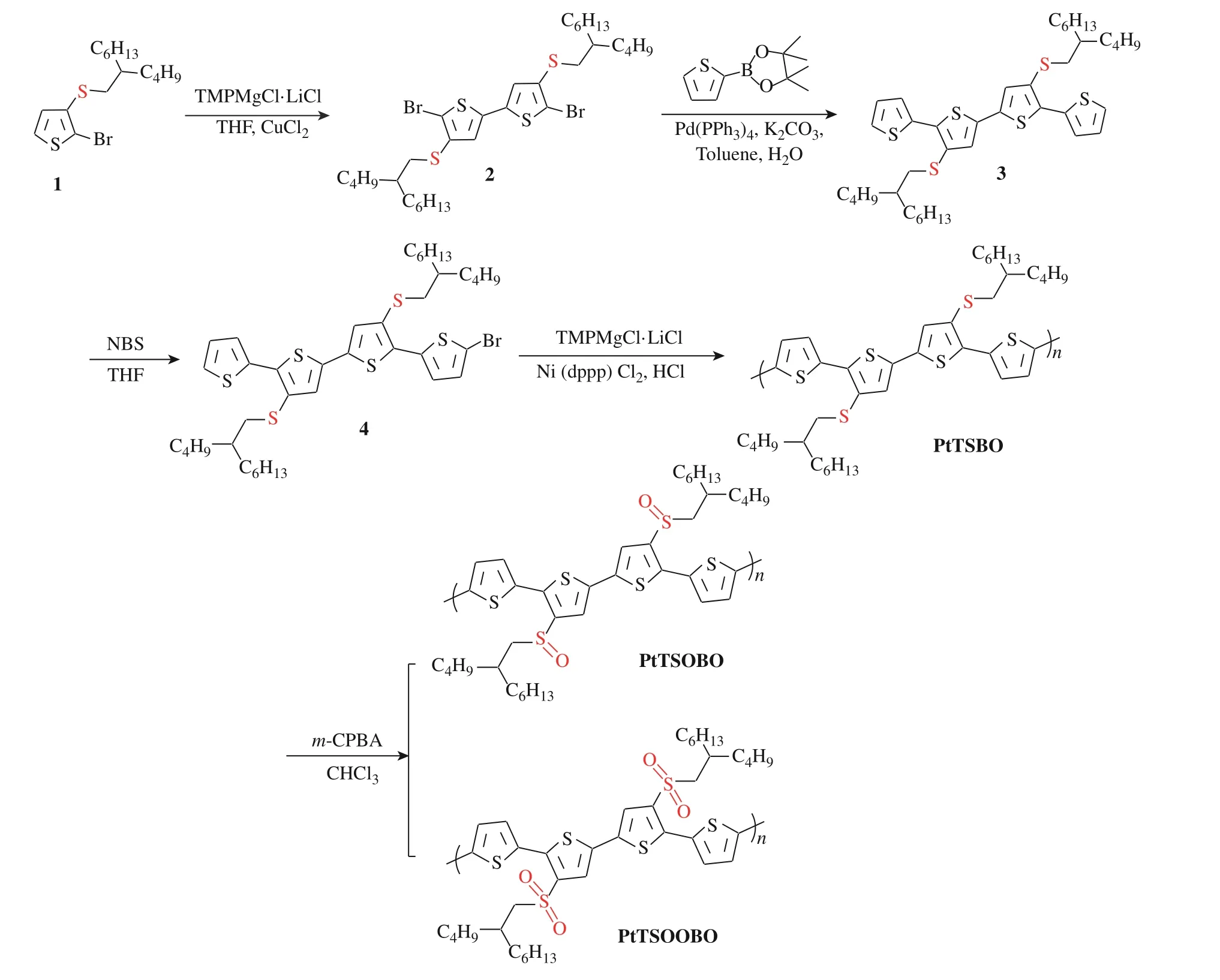
图1 PtTSBO、PtTSOBO、PtTSOOBO的合成路线Fig. 1 Synthetic route of PtTSBO、PtTSOBO and PtTSOOBO
取烘干的200 mL Schlenk瓶,抽换气3次,加入化合物1(3.39 g, 9.40 mmol),快速抽换气3次,用注射器加入80 mL无水四氢呋喃搅拌溶解。将反应体系放入低温恒温反应器中,降温至-20 ℃。向反应体系中缓慢滴加1 mol/L TMPMgCl·LiCl溶液(10.5 mL, 10.5 mmol)后,缓慢恢复至室温搅拌1 h,再降温至-20 ℃,保持氩气氛围,快速加入无水氯化铜(1.30 g, 9.40 mmol),缓慢恢复至室温搅拌过夜。待反应结束,加50 mL去离子水淬灭反应,先将浅绿色不溶固体过滤,再用石油醚(3×100 mL)萃取,有机相经无水硫酸钠干燥;粗产物用硅胶柱纯化,石油醚作为洗脱剂,得到无色油状产物(标记为2,2.21 g, 产率为65%)。
1H-NMR(400 MHz, CDCl3)δ: 6.90 (s, 2H), 2.85 (d, 4H), 1.58~1.52 (m, 2H), 1.46~1.19 (m, 32H),0.92~0.84 (m, 12H)。
取烘干的200 mL Schlenk瓶,抽换气3次,加入化合物2(2.22 g, 3.06 mmol)、2-噻吩硼酸频哪醇酯(1.52 g,6.12 mmol)和碳酸钾(1.69 g, 12.24 mmol),快速抽换气3次,保持氩气氛围,加入Pd(PPh3)4(210 mg, 摩尔分数为3%),抽换气3次,然后加入60 mL甲苯和15 mL去离子水。在110 ℃下搅拌过夜后,将混合物冷却至室温,加入20 mL去离子水淬灭反应并用石油醚(3×100 mL)萃取。有机相经无水硫酸钠干燥;粗产物通过硅胶柱纯化,石油醚作为洗脱剂,得到淡黄色油状产物(标记为3,1.03 g, 产率为46%)。
1H-NMR (400 MHz, CDCl3)δ:7.39 (d,J= 4.7 Hz, 2H), 7.33 (d,J= 5.1 Hz, 2H), 7.10 (s, 2H), 7.06 (dd,J=5.1, 3.7 Hz, 2H), 2.90~2.81 (d, 4H), 1.81~1.75 (m, 2H), 1.54~1.16 (m, 32H), 0.88~0.76 (m, 12H);13C-NMR(100 MHz, CDCl3)δ:135.23, 134.66, 133.46, 129.41, 128.30, 126.98, 126.22, 126.17, 41.04, 38.09, 33.11, 32.83,31.89, 29.61, 28.82, 26.93, 26.58, 22.98, 22.71, 14.14。m/z(MALDI-TOF MS, C40H58S6):计算值为730.29;实际值为730.29。
取烘干的100 mL三口瓶,加入化合物3(900 mg, 1.23 mmol),用50 mL四氢呋喃搅拌溶解,在-10 ℃下加入NBS(219 mg, 1.23 mmol),保持低温避光反应3 h,加入亚硫酸钠水溶液淬灭反应,用二氯甲烷(3×20 mL)萃取,有机相经无水硫酸钠干燥,粗产物用硅胶柱纯化,石油醚作为洗脱剂,得到淡黄色油状产物(标记为4,328 mg, 产率为33%)。
1H-NMR(400 MHz, CDCl3)δ:7.27 (s, 1H), 7.26 (s, 1H), 7.18 (dd,J= 3.6, 1.1 Hz, 1H), 7.13 (d,J= 4.0 Hz,1H), 7.10 (s, 1H), 7.03 (dd,J= 5.1, 3.6 Hz, 1H), 6.99 (d,J= 4.0 Hz, 1H), 2.88~2.83 (m, 4H), 1.86~1.75 (m,1H), 1.77~1.71 (m, 1H), 1.65~1.06 (m, 32H), 0.88~0.76 (m, 12H);13C-NMR (100 MHz, CDCl3) :137.05, 136.27,135.56, 134.91, 133.43, 132.89, 130.47, 129.97, 129.44, 128.86, 128.45, 127.98, 125.72, 125.09, 124.15, 113.50, 41.38,41.35, 38.15, 38.12, 34.67, 33.15, 33.12, 32.87, 32.85, 31.89, 31.59, 29.64, 29.06, 28.84, 26.92, 26.61, 25.28, 22.99,22.66, 22.61, 20.70, 20.45, 11.43。m/z(MALDI-TOF MS, C40H57S6Br):计算值为810.16;实际值为810.31。
取烘干的25 mL Schlenk瓶,抽换气3次,加入化合物4(200 mg, 0.25 mmol),抽换气3次,加入10 mL无水四氢呋喃;冷却至0 ℃后,向反应体系中滴加1 mol/L TMPMgCl·LiCl溶液(0.30 mL, 0.30 mmol),缓慢恢复到室温搅拌1 h;将溶液加热至35 ℃,在氩气氛围下快速加入Ni(dppp)Cl2催化剂(9 mg,0.013 mmol),再在50 ℃下搅拌5 h;待反应结束,将深红色溶液冷却至室温,用5 mol/L盐酸淬灭,并沉降到甲醇中,真空抽滤后得到粗产物。将粗产物倒入滤纸筒内,依次使用甲醇、正己烷、氯仿索氏提取,浓缩氯仿相得到紫色固体状的聚合物PtTSBO(80 mg, 产率为44%)。
1H-NMR(400 MHz, C2D2Cl4)δ:7.33 (s, 2H), 7.10 (s, 4H), 2.89 (m, 4H), 1.68~1.56 (m, 2H), 1.52~1.19 (m,32H), 0.92~0.80 (m, 12H)。GPC:Mn= 1.65×104,Mw= 2.36×104,Đ= 1.43。C40H56S6的元素分析计算值为:wC,65.88%;wH, 7.74%;wS, 26.38%; 实际值为:wC, 64.93%;wH, 7.57%;wS, 25.97%。
1.3.2 聚合物PtTSOBO、PtTSOOBO的合成 取烘干的100 mL两口瓶,抽换气3次,加入聚合物PtTSBO(82 mg, 重复单元0.11 mmol),抽换气3次,加入50 mL无水氯仿搅拌溶解,冰浴下加入m-CPBA(60 mg,0.22 mmol),低温下反应20 min。待反应结束后,用旋转蒸发仪减压旋蒸后得到深紫色的粗产物。将粗产物用适量氯仿溶解,沉降到乙醇中,真空抽滤后得到产物PtTSOBO(81 mg, 产率为98%)。
1H-NMR(400 MHz, C2D2Cl4)δ:7.64 (m, 2H), 7.18 (m, 4H), 3.00 (m, 2H), 2.81 (m, 2H), 2.03~1.94 (m,2H), 1.60~1.15 (m, 32H), 0.95~0.80 (m, 12H)。 C40H56S6O2的元素分析计算值为:wC, 63.11%;wH, 7.42%;wS,25.27%; 实际值为:wC, 62.32%;wH, 7.53%;wS, 24.92%。
聚合物PtTSOOBO的合成方法同上。加入聚合物PtTSBO(110 mg, 重复单元0.15 mmol)后,低温下反应1 h,真空抽滤后得到产物PtTSOOBO(109 mg, 产率为99%)。
1H-NMR (400 MHz, C2D2Cl4)δ:7.66~7.47 (m, 4H), 7.23~7.15 (m, 2H), 3.05 (m, 4H), 2.05~1.89 (m,2H), 1.64~1.07 (m, 32H), 0.91~0.75 (m, 12H)。C40H56S6O4的元素分析计算值为:wC, 60.57%;wH, 7.12%;wS,24.25%; 实际值为:wC, 60.32%;wH, 7.37%;wS, 24.77%。
2 结果与讨论
2.1 聚噻吩材料的合成
通常适用于KCTP法的单体共轭长度是受限的,但当单体共轭区域过大时,催化剂转移的效率和选择性将显著降低[10]。本文的单体由4个噻吩单元组成,并含有硫醚等容易使催化剂中毒的基团[18],对聚合反应提出了较大的挑战。参照Noonan课题组[19]的方法,采用非亲核性Knochel-Hauser碱试剂TMPMgCl·LiCl与单溴代的噻吩四聚体4进行反应,充分保证了每个单体都形成格氏试剂,加入催化剂Ni(dppp)Cl2,在50 ℃下搅拌反应5 h,成功得到了聚合物PtTSBO。聚合物的结构通过核磁共振氢谱、元素分析和凝胶渗透色谱进行了确认。PtTSBO聚合的成功合成意味着KCTP法在共轭聚合物领域依旧有着广泛的发展空间。
为了探索后修饰策略改性聚噻吩的可行性,本文从以下3个方面进行着手:(1)寻找合适的条件来改性聚合物(溶剂、温度、m-CPBA添加量);(2)确定所需的反应时间;(3)避免副反应的发生(主链中存在着可能易于氧化的噻吩环)。首先,将聚合物PtTSBO用溶剂氯仿溶解,使用m-CPBA对其进行氧化。同时,为了避免噻吩环上的硫原子被氧化[20],控制了反应温度。经过反复尝试,为了生成PtTSOBO,需要加入PtTSBO重复单元2.0倍物质的量的m-CPBA,而生成PtTSOOBO需要加入4.2倍物质的量的m-CPBA,反应路线见图1。根据实验可得,m-CPBA对硫代烷基侧链在低温下依旧具有良好的氧化效果,并且反应速率极快,加入氧化剂20 min后,侧链上的硫醚全部转化。待反应结束后,将产物用适量氯仿溶解,然后沉降到乙醇中,通过过滤获得改性后的聚合物,而副产物间氯苯甲酸溶解在乙醇中被除去。
PtTSBO、PtTSOBO和PtTSOOBO的1H-NMR谱图如图2所示,随着引入强吸电子的亚砜和砜基,聚噻吩主链上的特征氢峰(Ha和Hb)均向低场移动,Hb从化学位移7.33分别偏移到了7.60和7.61,Ha从7.10偏移到了7.16和7.19。此外,在高场的烷基区,硫代烷基链上与硫原子相连的CH2特征峰(Hc)随着亚砜和砜基的引入,也逐渐向低场移动。同时,随着亚砜基的引入,Hc特征峰出现了裂分,这是亚砜基产生的手性致使2个氢原子的化学环境不同所造成的。同时,从PtTSOBO和PtTSOOBO的1H-NMR谱图分析得到,副产物间氯苯甲酸已经除去,而主链的噻吩环没有发生副反应。

图2 样品的1H-NMR谱图Fig. 2 1H-NMR spectra of samples
2.2 密度泛函理论计算
为了研究引入亚砜和砜基对聚合物的分子骨架构型以及平面性的影响,采用DFT模拟了3个重复单元的优势构型和电荷分布。
PtTSBO、PtTSOBO和PtTSOOBO的分子构型、联噻吩单元与侧链功能化的联噻吩单元之间的二面角和分子骨架的电子云分布如图3所示。结果显示,引入亚砜和砜基对PtTSBO的平面性有着重要影响。未进行后修饰改性的聚合物PtTSBO对应的二面角(11.20°)最小,分子主链平面性最高。与PtTSBO相比,亚砜基聚合物PtTSOBO的分子骨架逐渐扭曲,二面角增大到27.48°,减弱了主链的平面性。随着砜基的引入,聚合物PtTSOOBO的二面角进一步增大到38.17°,这是因为砜基具有较大的空间位阻,主链过大的扭曲将不利于分子堆积。
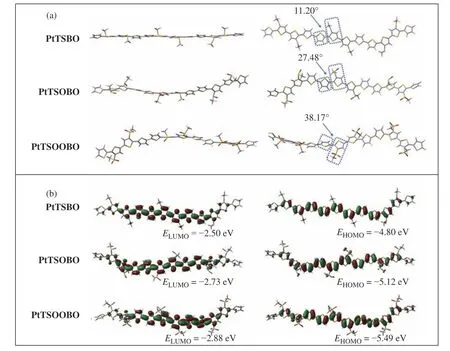
图3 PtTSBO、PtTSOBO和PtTSOOBO的(a)构型与(b)分子轨道示意图Fig. 3 (a) Schematic diagram of configuration and (b) molecular orbital of PtTSBO, PtTSOBO and PtTSOOBO
另一方面,这3种聚合物的HOMO和LUMO均在整个共轭主链上离域,但是强吸电子的亚砜和砜基会让聚合物共轭主链的LUMO离域程度降低,继而影响聚合物能级。通过计算得到了这3种聚合物的HOMO和LUMO能级,随着亚砜和砜基的引入,相应聚合物的HOMO与LUMO能级逐步降低。
2.3 光学和电化学性质
为了研究引入亚砜和砜基对聚合物吸收光谱的影响,测试了这3种聚合物在氯仿溶液(7×10-4mg/mL)和薄膜状态下的吸收光谱(图4),具体相关参数归纳在表1中。与PtTSBO相比,改性后的PtTSOBO在溶液中具有更高的摩尔消光系数(ε),说明引入亚砜基可以加强聚合物的吸光能力,但是砜基引入后,摩尔消光系数有所降低。在溶液状态下,PtTSOBO的最大吸收峰比PtTSBO蓝移了9 nm,但是两者在稀溶液状态下,均可以在长波方向观察到明显的肩峰,这说明即使在溶液里,也表现出了一定的聚集。但是引入砜基后,PtTSOOBO的最大吸收峰蓝移了89 nm,而且堆积峰消失,这与砜基的吸电子能力和立体位阻有关。在薄膜状态下,改性聚合物的吸收光谱呈现更大幅度的蓝移,其中,PtTSOOBO蓝移了129 nm,这是由于砜基具有较大的位阻效应,造成了分子骨架的平面性变差,不利于分子堆积。此外,聚合物PtTSBO和PtTSOBO的光学带隙相近,分别为1.87、1.90 eV,聚合物PtTSOOBO光学带隙超过了2.00 eV。

图4 聚合物在(a)氯仿溶液和(b)薄膜状态下的紫外-可见分光光谱Fig. 4 UV-Vis spectra of polymers in (a) chloroform solution and (b) film
为了研究聚合物的能级,通过循环伏安法扫描出聚合物的氧化电位,得到聚合物的HOMO能级(EHOMO),再由聚合物的光学带隙计算得出相应的LUMO能级(ELUMO),相关数据见图5以及表1。

图5 聚合物的(a)循环伏安曲线和(b)能级示意图Fig. 5 (a) Cyclic voltammetry curves and (b) schematic diagram of energy levels of polymers

表1 聚合物的光学和电化学性质Table 1 Optical and electrical properties of the polymers
显然,后修饰改性能使聚合物的HOMO和LUMO能级降低,这与DFT计算结果一致。另外,对于降低聚噻吩材料的HOMO能级而言,引入砜基的效果更为显著。
2.4 器件光伏性能与形貌
为了研究后修饰改性对聚噻吩光伏性能的影响,本文选用O-IDTBR(O-IDTBR:(5Z,5Z)-5,5-((7,7-(4,4,9,9-四辛基-4,9-二氢-S-引达省并[1,2-B:5,6-B]二噻吩基)双(苯并[C][1,2,5]噻二唑基))双(甲基亚甲基))双(3-乙基噻唑烷-2,4-二酮))作为受体材料[21,22],在同样的条件下制备了聚合物的光伏器件。器件结构为正置型器件:ITO/PEDOT:PSS/donor:O-IDTBR/ZnO/Al(PEDOT:聚(3,4-乙烯基二氧噻吩);PSS:聚(苯乙烯磺酸盐),聚合物与受体分子的质量比均为1∶1,体系加工溶剂为氯仿,使用氯萘(体积分数为0.5%)作为添加剂,110 ℃热退火5 min后性能最优,这3种聚合物的光伏器件性能参数见图6和表2。

图6 基于聚合物光伏器件的J-V曲线Fig. 6 J-V curves of devices based on the polymers
由表2可知,引入亚砜和砜基后,基于后修饰改性的聚合物光伏器件的Voc逐步提高,这与聚合物HOMO能级降低的趋势一致。PtTSOBO和PtTSOOBO具有大共轭体系、吸电子能力更强的联噻吩单元,基于PtTSOBO和PtTSOOBO的光伏器件,其Voc相对基于PtTSBO的光伏期间分别提升了0.40 V和0.46 V。但是,随着亚砜和砜基的引入,光伏器件的短路电流密度(Jsc)和填充因子(FF)却大幅度降低。

表2 基于给体与O-IDTBR器件的光伏性能Table 2 Photovolatic properties of devices based on donor:O-IDTBR
从PtTSBO体系的AFM高度图(图7(a~c))可以得到,随着亚砜和砜基的引入,共混膜的均方根粗糙度(RMS)值逐渐降低,从7.00 nm分别降低到了3.41 nm和1.13 nm,相分离程度逐渐减弱。基于PtTSOBO共混膜的相分离程度较差,而基于PtTSOOBO共混膜的表面平整,没有明显的相分离(图7(d~f))。

图7 最优条件下PtTSBO(a, d)、PtTSOBO(b, e)和PtTSOOBO(c, f)共混膜的AFM高度图(上)与相图(下)Fig. 7 AFM height (up) and phase (down) diagrams of blend films for PtTSBO (a, d), PtTSOBO (b, e) and PtTSOOBO (c, f) under optimal conditions
结合以上研究结果,改性后聚合物光伏性能降低的原因主要在于亚砜和砜基对聚合物构型的影响。PtTSOBO和PtTSOOBO的主链扭曲,平面性较差,不利于固态下分子之间的堆积,造成材料结晶性较差。与结晶性同样较低的小分子受体材料OIDTBR共混后,共混薄膜无法形成纯净度较高的自富集域,从而抑制了电荷的传输速率,并造成严重的载流子复合,最终使得器件的Jsc和FF降低。因此,对于后修饰改性的聚噻吩光伏器件还有待进一步研究和优化,比如选用能级、混溶性更加匹配的受体材料,或将改性后的聚噻吩作为电子受体材料进行光伏性能研究。
3 结 论
(1)利用Kumada催化剂转移缩聚法,成功实现了含有4个噻吩单元和硫醚侧链单体的聚合,得到了新型聚噻吩材料PtTSBO,拓宽了KTCP法的适用范围。
(2)通过控制反应温度和氧化剂用量,选择性地将聚合物PtTSBO的硫醚氧化成亚砜或砜,成功地在聚噻吩中引入了强吸电子基团,证明了后修饰策略在共轭聚合物改性上的有效性。
(3)利用后修饰策略对聚噻吩材料进行改性,提升了相应光伏器件的开路电压,但是降低了聚合物的平面性,不利于其固态堆积,从而影响了器件总体的光伏性能。
猜你喜欢
中学化学(2024年1期)2024-05-26 13:20:27
水产科学(2023年4期)2023-07-22 01:33:56
中国计算机报(2019年26期)2019-08-27 08:16:16
浙江化工(2017年11期)2017-12-19 06:11:46
中学生数理化·高二版(2017年2期)2017-04-19 16:29:54
中学化学(2016年12期)2017-02-05 17:24:23
当代化工研究(2016年1期)2016-03-16 22:00:24
合成化学(2015年10期)2016-01-17 08:56:47
浙江化工(2015年10期)2015-03-10 04:41:38
应用化工(2014年9期)2014-08-10 14:05:08
