
鳃耳肾谱系疾病2例病例报告
2021-10-22 07:09:36孙留玉苏白鸽
中国循证儿科杂志 2021年4期
孙留玉 钟 贞 朱 颖 苏白鸽 丁 洁 王 芳
1 病例资料
例1:男,汉族,44 d。主因胎儿期发现双肾小、血肌酐升高43 d入北京大学第一医院(我院)儿科肾脏病房。患儿系G1P1,母亲孕期体健、规律产检,孕38+5周时胎儿超声示双肾小(右肾长径2.3 cm~2.6 cm,左肾长径2.1 cm~2.5 cm)。患儿于母孕39+1周顺产娩出。43 d前查血肌酐为155.2 μmol·L-1,41 d前复查为191.2 μmol·L-1。43 d前泌尿系超声示双肾小(右肾长2.6 cm、左肾长2.1 cm)、双肾多发囊肿。为进一步明确病因来我院。新生儿听力筛查未通过。父母体健,否认近亲结婚,否认类似病史。外祖母60余岁诊断囊性肾脏病,余家族成员否认相关疾病家族史。
入院查体:BP (70~80)/(25~40)mmHg,体重 4 920 g (P50~P75),身长54 cm (P50),头围 37.5 cm (P50~P75)。右耳小耳畸形、外耳道闭锁,双侧耳前瘘管,双侧颈部可见瘘口,未见明显分泌物(图1),心、肺、腹查体未见异常,神经系统查体示竖头稳,可追光和追物,但追声稍差。
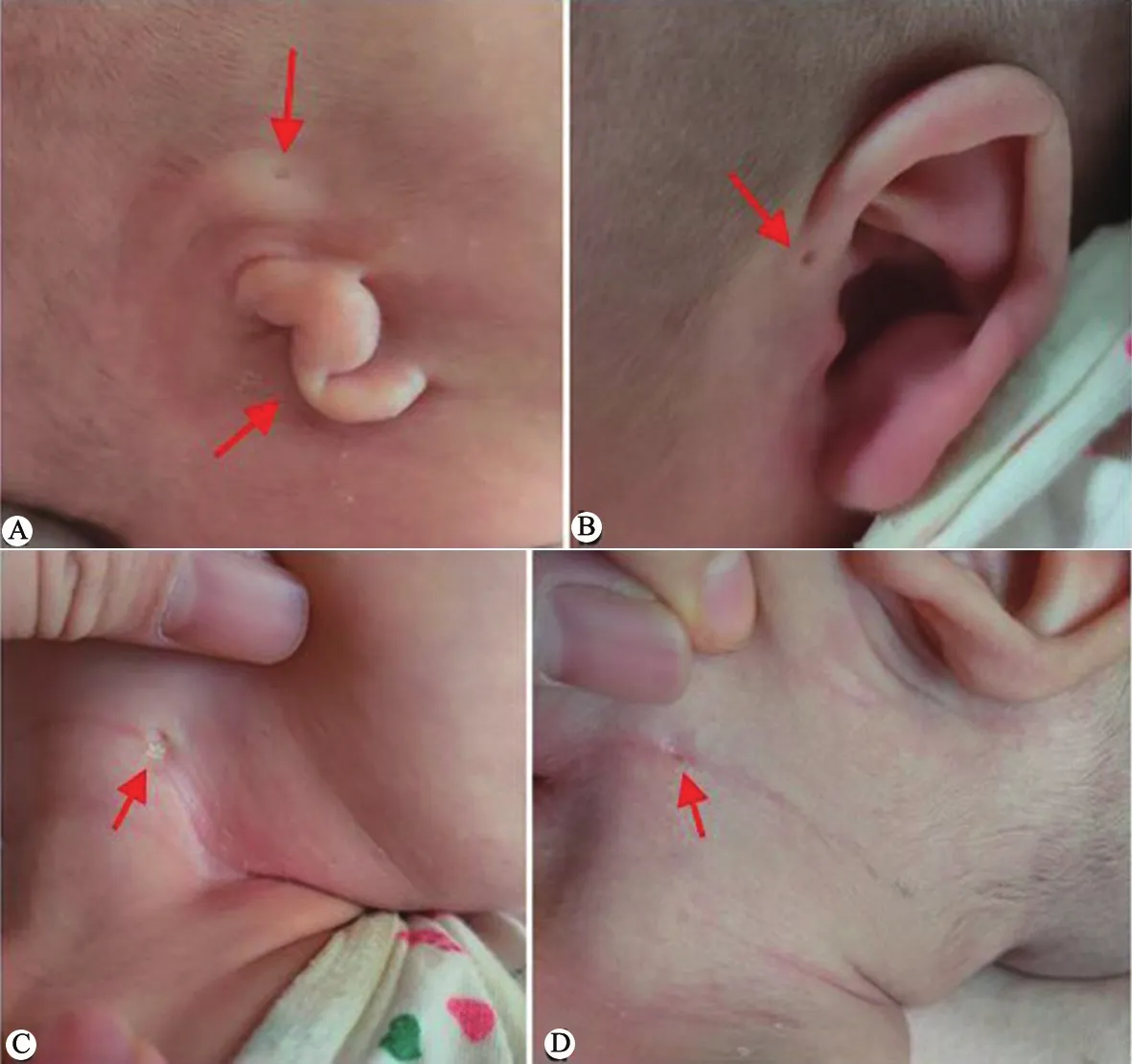
图1 例1耳部畸形和鳃裂瘘
入院后辅助检查:血常规示Hb 118 g·L-1;尿常规示比重1.002,蛋白和尿糖均阴性,镜检无RBC和WBC;血生化示肌酐 97.4 μmol·L-1,BUN 7.23 mmol·L-1;尿蛋白/尿肌酐 1.11;尿微量白蛋白/肌酐 49.1 mg·g-1、尿α1微球蛋白47.8(参考范围:0~12)mg·L-1;尿蛋白电泳显示小分子蛋白77.6%、白蛋白22.4%。听觉脑干诱发电位阈值左耳气导60 dBnHL、骨导30 dBnHL,右耳骨导35 dBnHL;左耳多频稳态示500、1 000、2 000和4 000 Hz分别为40、30、20和20 dBnHL;左耳鼓室图检查示单峰,提示右耳传导性耳聋、左耳听力正常。腹部超声示双肾小(右肾长径3.1 cm,左肾长径2.7 cm)、双肾囊肿;颞骨CT示右耳小耳畸形,右侧外耳道闭锁,右侧中耳及双侧内耳发育畸形(图2)。临床诊断鳃耳肾谱系疾病(BORSD),采用目标区域捕获二代测序(深圳华大临床检验中心)进行鳃耳肾综合征(BORS)致病基因检测分析,异常结果经Sanger测序验证。结果提示患儿EYA1基因 (NM_000503.4)10号外显子杂合无义变异c.889C>T(p.Arg297*),其父母未检测到相同变异。该变异已被报道[1],在dbSNP数据库、千人亚洲数据库、ESP6500数据库和ExAC数据库无记录。第297位Arg在脊椎动物中高度保守。根据美国医学遗传学与基因组学学会(ACMG)遗传变异分类标准指南,该变异为致病变异(PVS1 PS1 PS2)。ClinVar数据库评价该变异亦为致病变异。
例2:男,维吾尔族,7.6岁。因“贫血、血肌酐升高2.5年,加重1月余”入住我院儿科肾脏病房。2.5年前因说话发音不准确就诊于当地医院,检查示贫血(Hb 109 g·L-1)、肾功能减退(血肌酐 83.9 μmol·L-1、BUN 7.8 mmol·L-1)和低比重尿(尿比重1.010),未予进一步诊治。1月余前患“肺炎”后出现双眼睑水肿,伴泡沫尿和夜尿增多,当地医院检查示贫血(Hb 72 g·L-1)、肾功能衰竭[血肌酐 307.1 μmol·L-1、BUN 29.3 mmol·L-1,肾动态显像法示总肾小球滤过率7.54(左肾3.19、右肾4.35)mL·min-1、低比重尿(尿比重1.004)和肾病水平蛋白尿(24 h尿蛋白2.07 g),为进一步明确病因来我院。患儿自发病以来食欲可,夜尿增多,大便正常。家长诉患儿听力降低。患儿系G3P3,母亲孕期体健、未规律产检,足月顺产。患儿体格及精神神经发育与正常同龄儿大致相同。现上小学一年级,学习成绩一般。父母体健,否认近亲结婚和家族类似病史。

图2 例1颞骨CT
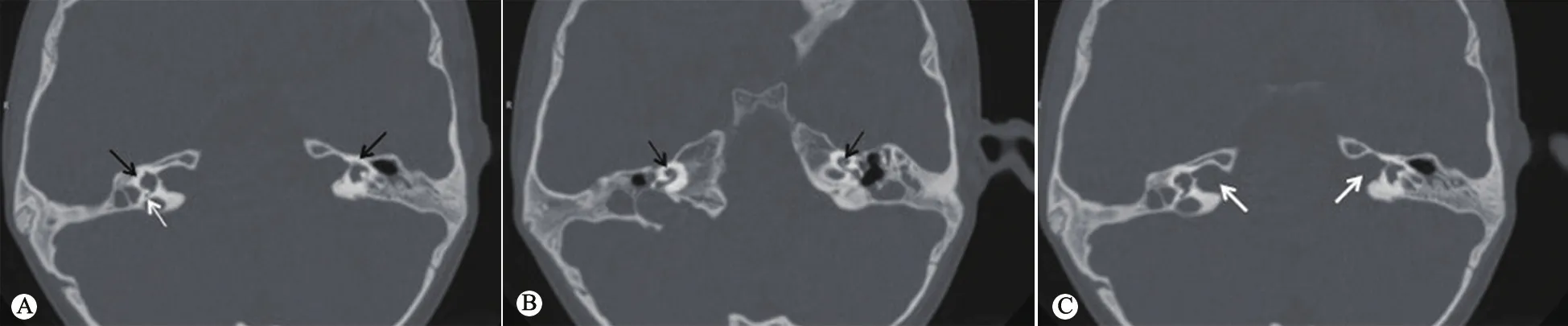
入院查体:BP 128/80 mmHg,身高123 cm(P10~P25),体重19 kg( 入院后辅助检查:血常规示Hb 86 g·L-1,平均红细胞体积85.6 fL、平均红细胞血红蛋白含量 28.4 pg、平均红细胞血红蛋白浓度332 g·L-1;尿常规示蛋白3+、比重1.010、尿糖阴性、镜检RBC 0~2个/HP;血生化示肌酐327.5 μmol·L-1、BUN 29.1 mmol·L-1、ALB 36.0 g·L-1、胆固醇5.6 mmol·L-1、碳酸氢根16.5 mmol·L-1、钙2.1 mmol·L-1、磷2.20 mmol·L-1;24 h尿蛋白定量2.58 g(每kg体重135.8mg,肾病水平);尿蛋白电泳示白蛋白66.8%、大分子蛋白27.9%、小分子蛋白5.3%;24 h内生肌酐清除率11.4 mL·min-1·1.73m-2;贫血筛查示转铁蛋白饱和度、铁蛋白、叶酸和维生素B12均正常;全段甲状旁腺素254.5(参考范围15~65)pg·mL-1;24 h动态血压示平均收缩压及舒张压均升高,昼夜收缩压、舒张压、负荷均升高,昼夜节律消失;超声心动图示室间隔轻度肥厚,主动脉少量反流,三尖瓣少量反流;泌尿系超声示双肾小(右肾长径4.1 cm、左肾长径6.1 cm),肾实质回声显著增强,皮髓质分界不清,左肾血流充盈不良,右肾内未见血流信号。听力测试示双耳混合性聋(图3),声导抗双耳B型曲线。颞骨CT示双侧中耳及内耳畸形(图4)。耳鼻喉科建议双耳佩戴助听器。8岁2个月开始规律腹膜透析治疗。临床诊断BORSD,采用目标区域捕获二代测序(深圳华大临床检验中心)进行该病致病基因检测分析,异常结果经Sanger测序验证。结果提示患儿EYA1基因外显子10杂合无义变异c.922C>T(p.Arg308*),其父母未检测到相同变异。该变异已被报道[1,5,11],在dbSNP数据库、千人亚洲数据库、ESP6500数据库和ExAC数据库无记录。根据ACMG遗传变异分类标准指南,该变异为致病变异(PVS1 PS1 PS2)。ClinVar数据库评价该变异亦为致病变异。 图3 例2听力测试结果 图4 例2颞骨CT 鳃-耳-肾综合征(BORS)是一种罕见的常染色体显性遗传性疾病,主要临床表现为耳部畸形(包括外耳、中耳和内耳结构畸形)、耳聋(可呈传导性、感音神经性或混合性)、鳃裂瘘、鳃裂囊肿和肾脏尿路畸形。缺乏肾脏尿路畸形时则称为鳃-耳综合征。研究显示,同一家系中不同患者可表现为BORS,亦可表现为鳃-耳综合征[1],因此近来认为这两种疾病是同一谱系疾病的不同表现,将其统称为BORSD[2]。中国BORSD的临床诊断病例报道始见于1997年[3],分子诊断病例报道始见于2015年[4]。国内已报道病例中,有肾脏异常者不多[3, 5-7],低于国外报道[8-10]。终末期肾病是影响BORSD预后的一个重要因素[2,11]。 BORSD核心症状包括:①鳃裂瘘管或囊肿,②耳聋,③耳前瘘管,④肾脏畸形(肾脏缺如、发育不全、发育不良,肾盂输尿管连接部梗阻,肾盏憩室或囊肿,肾盏、肾盂扩张,肾盂积水,膀胱输尿管反流等)。次要症状包括:①外耳异常(耳前凸起、杯状耳、副耳郭,外耳道闭锁或狭窄等),②中耳畸形(听骨链畸形、错位、脱臼或固定,鼓室狭窄或畸形等),③内耳异常(耳蜗发育不全、耳蜗及前庭导管扩张、外半规管发育不全等),④其他,如面部不对称、腭畸形等。符合下列标准中任意一条者即可临床诊断BORS[2]:≥3项核心症状;2项核心症状+2项次要症状;1项核心症状+1名家系成员患BORSD。 本文2例均因肾功能减退就诊,泌尿系超声显示小肾脏,考虑肾功能减退为慢性,病因需考虑双肾发育不良或不全。体格检查发现鳃裂瘘管和耳前瘘管,例1尚有右耳小耳畸形和外耳道闭锁,故临床考虑BORSD,进而予以相关的辅助检查,检测到耳部畸形和耳聋,遗传学检测分析显示EYA1基因存在无义突变,明确诊断为BORSD,为后续合理管理患儿和客观正确的遗传咨询奠定了基础。 并非所有BORSD患儿均完成泌尿系影像学检查,因此肾脏尿路畸形的确切发生率并不清楚。研究显示[8-10],BORS肾脏尿路畸形检出率>60%,其中以肾发育不全/不良最常见。此外,尚有研究报道肾小球病变。Morisada等[11]报道了1例EYA1基因缺失突变所致的BORS肾活检病理为膜性肾病;Gigante等[12]报道了1例EYA1基因剪接突变所致BORS表现包括非肾病水平蛋白尿,肾活检病理为局灶节段肾小球硬化。本文2例均表现为小肾脏,虽未行肾活检组织病理学检查,但结合其临床表现,推测例1为双肾发育不良可能性大,例2为双肾发育不全可能性大:例1胎儿期便发现肾脏小,生后第2 d发现肾功能减退,且有肾小管性蛋白尿和双肾囊肿,提示肾实质存在发育不良;例2于5岁2个月发现肾功能下降,7岁7个月进展至终末期肾病,同时尚表现有肾病水平的肾小球性蛋白尿,提示肾实质具有结构正常的肾单位,但是正常肾单位数量少,随疾病进展最终出现不可逆的肾功能下降。 已知终末期肾病是影响BORSD预后的一个重要因素[2,11],因而正确评价BORSD患儿肾功能具有重要意义。临床实践中血肌酐因检测方便且经济,成为间接评价肾脏功能应用最为广泛的指标。然而,值得注意的是,血肌酐水平受肌肉容积影响,儿童血肌酐参考范围随年龄不同而不同[13],若依据血生化检查报告中提供的基于成人的血肌酐参考范围判断BORSD患儿的肾功能,易造成误判。本文例2于5岁2个月就诊时查血肌酐83.9 mol·L-1,高于同年龄参考区间上限高值57.7 mol·L-1[13],表明已发生肾功能减退,应及时转诊至儿科肾脏专科以明确诊断并接受治疗,遗憾的是该患儿发展至终末期肾病时才考虑转诊,丧失了延缓肾功能进展的治疗机会。因此,临床医生除了关注BORSD的鳃裂畸形、耳部畸形和耳聋外,需行肾脏专科会诊进行肾脏评估。 BORSD基因型和肾脏表型间尚未发现相关性,且同一家系不同患者肾脏表型可不相同[1,10]。以EYA1基因的基因型p.Arg297*和p.Arg308*为例,已有报道显示患者可无肾脏异常[1,4],而本文2例患儿均表现为小肾脏和终末期肾病。此外,BORSD各年龄均可发生肾衰竭,亦可早在胎儿期便发生肾功能衰竭,引起羊水过少[9-10]。 本文所有作者均声明不存在利益冲突。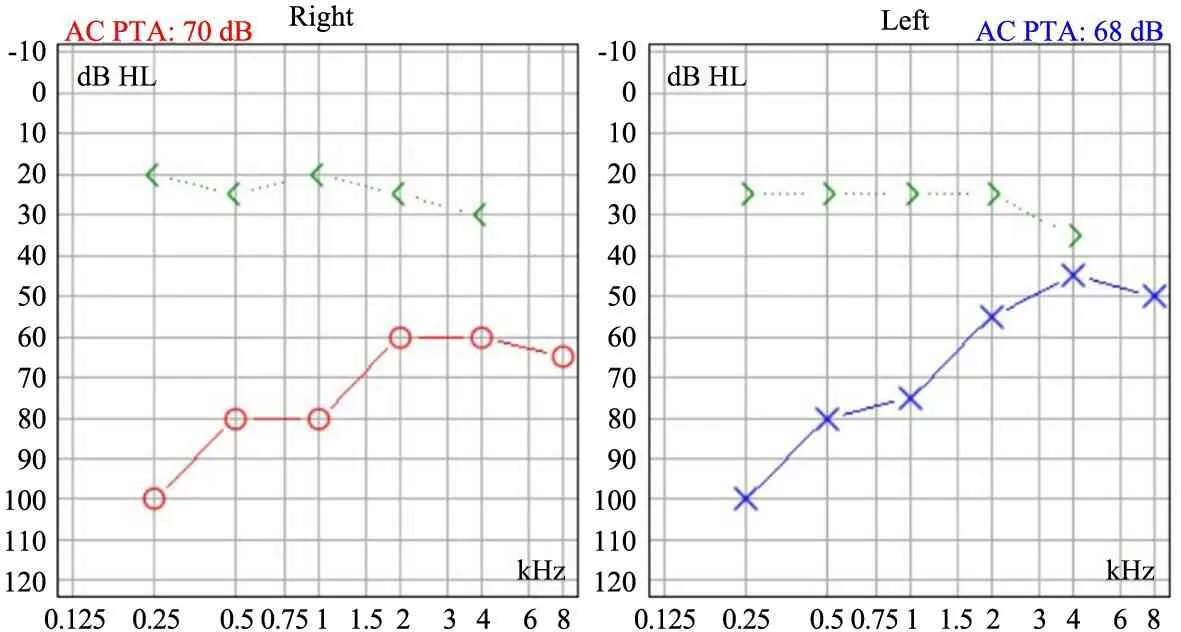
2 讨论
猜你喜欢
中国医学影像技术(2022年12期)2022-12-28 09:46:48
蚌埠医学院学报(2019年9期)2019-10-14 07:00:44
新农业(2019年24期)2019-01-06 07:14:32
浙江医学(2018年16期)2018-09-08 05:58:00
解剖学杂志(2018年2期)2018-05-21 10:01:13
介入放射学杂志(2018年4期)2018-04-18 00:56:50
长春中医药大学学报(2017年1期)2017-04-16 05:56:52
哈尔滨医药(2015年5期)2015-12-01 03:58:09
医学研究杂志(2015年4期)2015-06-10 06:42:43
家庭医学·下半月(2014年7期)2014-03-18 22:28:48
