
13号染色体q33.2-q34缺失伴慢性肾脏病1例病例报告
2021-10-22 07:09:40冯春月沈辉君傅海东毛建华
中国循证儿科杂志 2021年4期
冯春月 刘 飞 沈辉君 傅海东 毛建华
1 病例资料
女,12岁2月,因“发热伴咳嗽9 d,胃纳差5 d”于2021年2月收治于浙江大学医学院附属儿童医院(我院)。患儿9 d前出现发热(体温最高38.5℃,热型不详,物理降温后体温可降至正常),伴单声咳嗽,随之出现胃纳减退,进食后呕吐,呕吐物为胃内容物,逐渐出现拒食,仅喝水。
患儿系G1P1,因宫内缺氧于孕38周剖宫产娩出,出生体重2 kg,Apgar评分不详。生长发育落后于正常同龄儿,半岁左右会抬头,2岁左右会走路,可独立行走;9岁以后走路不稳,需扶走;自幼语言发育落后,耳聋,能发声但口齿不清,能用肢体动作与家人交流;智力水平明显低下。患儿父母平素体健,否认近亲结婚,否认遗传性疾病家族史。母亲否认孕期服用药物、酗酒、接受放射性检查等。患儿胞弟6岁,体健。
既往史:患儿生后即诊断“先天性肛门闭锁,直肠会阴瘘”,3月龄至1岁6个月期间先后予结肠造瘘术、肛门成形术、肠造瘘回纳术。生后心脏听诊发现胸骨左缘2~3肋间3/6级柔和杂音,P2分裂;心脏超声显示房间隔缺损(继发孔直径2.1 cm),冠状静脉窦扩大,左上腔静脉残存;1岁9个月时全麻体外循环下行房间隔缺损修补术。听力筛查双耳均未通过,4岁时行人工耳蜗植入术。1岁6月时尿常规显示尿蛋白++,尿红细胞85·μL-1,尿比重1.015;血肌酐31 μmol·L-1;肝胆胰脾超声未见异常;泌尿系超声双肾形态未见异常,包膜光滑,皮质回声均匀,肾盂不扩张;头颅CT显示前额稍尖,脑结构未见异常。4岁半时行染色体核型分析(图1A):46,XX,del(13)(q33);基因芯片分析(Affymetrix CytoScan HD Array,图1B)提示13q33.2-q34发生缺失,缺失片段大小约8.8 Mb,涉及OMIM基因共27个,包括EFNB2、ARGLU1、LIG4、SLC10A2、TNFSF13B、IRS2、COL4A1、COL4A2、CARS2、ING1、ARHGEF7、SOX1、ATP11A、MCF2L、F7、F10、PROZ、PCID2、CUL4A、LAMP1、ADPRHL1、TFDP1、ATP4B、GRK1、GAS6、RASA3、UPF3A。

图1 患儿染色体核型图和基因芯片检测结果
体格检查:T 37.5℃,P 112次/min,R 24次/min,BP 106/48mmHg。神志欠清,瞳孔等大等圆,对光反射灵敏,呼之反应迟钝,表情淡漠,精神反应欠佳,颈抵抗。身长115 cm(<-3SD),体重15 kg(<-3SD),头围47 cm。小头畸形、眼距宽、鼻梁扁平,面色苍白,皮肤、巩膜无黄染,皮肤干燥,弹性差,皮下脂肪薄,胸部正中线可见长约8 cm手术瘢痕,上腹部可见长约5 cm手术瘢痕。两肺呼吸音粗,未闻及啰音,心律齐,心前区未闻及杂音腹软,无压痛,肝脾肋下未及。四肢肌张力高,肌力正常,四肢屈曲,双足内收,双侧巴氏征阳性,踝阵挛阳性。乳房未发育,未见腋毛;女性会阴,未见阴毛,肛周可见陈旧性手术瘢痕。
辅助检查:血常规WBC 14.4×109·L-1,Hb 75 g·L-1,PLT 425×109·L-1,CRP<0.5 mg·L-1。血气和电解质分析:pH 7.198, K+2.5 mmol·L-1, Na+125 mmol·L-1,HCO3-9.1 mmol·L-1,实际碱剩余(ABE)
-17.6 mmol·L-1, 标准碱剩余(SBE)-17.5mmol·L-1。血生化显示钙1.58(参考值:2.20~2.65) mmol·L-1,磷1.01(参考值:1.29~2.26) mmol·L-1,肌酐258 μmol·L-1,尿素 28.7 mmol·L-1,尿酸652 μmol·L-1, 胱抑素C 2.03 mg·L-1,eGFR 16.2 mL·min-1·1.73 m-2。甲状旁腺素437.4(参考值:<78.4)pg·mL-1。 免疫球蛋白和补体、凝血谱、血氨、促红细胞生成素、抗核抗体20项、T细胞亚群检测、乙肝5项定性、HIV、梅毒螺旋体、丙肝、脑脊液常规、脑脊液生化及培养均未见异常。尿常规显示,尿蛋白+,尿红细胞镜检4~11个/HP。24 h尿蛋白定量1 218.7 mg。尿微量白蛋白102.8(参考值0~20) mg·L-1,尿α1微球蛋白118.6(参考值0~12)mg·L-1,尿β2微球蛋白 31.4(参考值0~0.3)mg·L-1。泌尿系B超显示,左肾大小6.4 cm×3.1 cm,右肾大小6.5 cm×2.6 cm,皮质回声明显增强,皮髓质分界不清;诊断“双肾外形偏小,双肾弥漫性病变”;左肾静脉未见明显受压。心脏彩超示,冠状静脉窦扩大,左上腔静脉残存,三尖瓣轻度反流。肝胆胰脾B超未见异常。X线胸片显示,两肺野模糊,胸骨和肋骨骨质疏松,肋骨前端稍膨大,右第1~4后肋、左第2~4后肋透亮线影。心电图显示,窦性心动过速,Ⅰ度房室传导阻滞,Q-T间期延长,ST段改变。头颅CT示脑沟裂宽深。
诊治经过:入院诊断为慢性肾脏病(CKD)4期,急性支气管肺炎,代谢性酸中毒,电解质紊乱,中度贫血,染色体异常,脑发育落后,生长发育迟缓,肛门闭锁术后,先天性心脏病术后。予补钾补液、抗感染、纠酸、补钙及活性维生素D、补铁及促红细胞生成素等对症支持,患儿体温恢复正常,咳嗽减少,胃纳好转。代谢性酸中毒、低钾血症、贫血有所改善,血尿、蛋白尿及肾损伤无明显改善。入院第13天复查血常规:WBC 5.8×109·L-1,Hb 90 g·L-1,PLT 146×109·L-1,CRP<0.5 mg·L-1。血气分析PH 7.387, K+3.3 mmol·L-1, Na+137 mmol·L-1,HCO3-18.3 mmol·L-1,ABE -5.4 mmol·L-1, SBE -5.1 mmol·L-1, Lac 1.1 mmol·L-1。血生化显示,钙1.95 mmol·L-1,磷0.40 mmol·L-1,镁0.78 mmol·L-1,肌酐237 μmol·L-1,尿素 10.2 mmol·L-1,尿酸229 μmol·L-1, 胱抑素C 2.36 mg·L-1。尿常规显示,尿蛋白+,尿红细胞镜检4个/HP。每日尿量约600 mL,血压稳定,予出院。
出院半年时随访,患儿食欲可,每日尿量600~800 mL,血压正常,尿蛋白+,尿红细胞镜检5个/HP;血常规Hb 95 g·L-1,血肌酐242 μmol·L-1。
长期治疗计划:因患儿存在持续蛋白尿、生长发育迟缓、中度贫血、钙磷代谢紊乱等慢性肾脏病的常见并发症且控制不佳,建议长期避免应用肾毒性药物,监测血压、尿量,口服骨化三醇及钙剂以改善肾性骨病,应用铁剂及促红细胞生成素以维持Hb 110~120 g·L-1,定期监测心肺功能,CKD进入5期后需肾脏替代治疗,首选腹膜透析和血液透析。
2 讨论
13q缺失综合征是由于13号染色体长臂部分缺失导致的严重综合征[1]。早在1969年Allderdice等[2]即报告“13号染色体缺失综合征”,该病临床表型多样,根据遗传物质缺失的部位和大小可分为3种类型。①13q32带附近染色体区域缺失:一般表现为轻到中度的智力低下和面部畸形;②13q32带缺失:表型最严重,通常包括脑、眼、远端肢体、泌尿生殖道和胃肠道的畸形,以及严重的智力低下和身材矮小;③13q33-q34带远端缺失:多数表现为智力低下、小头畸形和男性生殖器畸形,一般不伴随其他严重畸形[3-5]。
本文患儿为13q33.2-q34缺失,除生长发育迟缓、特殊面容、生殖器畸形、听力异常、心脏异常外[6,7],生后不久尿检即有蛋白尿及血尿,当时肾功能及肾脏大小无异常,此后未定期复查,12岁就诊时已为CKD 4期。Sagi-Dain等[1]总结了60例单纯13号染色体q33.1-q34缺失的相关临床表型。81.7%的患儿有发育迟缓和/或智力低下,约50%有面部畸形和小头畸形,约20%出现抽搐,心血管畸形、身材矮小和肌张力减退各占10%~30%,约10%出现泌尿生殖系统畸形。13号染色体上存在许多关键基因,但由于目前已报道的病例数量有限和外显率水平不同,致病基因尚未完全确定。表1根据目前已有文献的报道情况,总结了13号染色体q33-q34缺失相关的基因及功能。
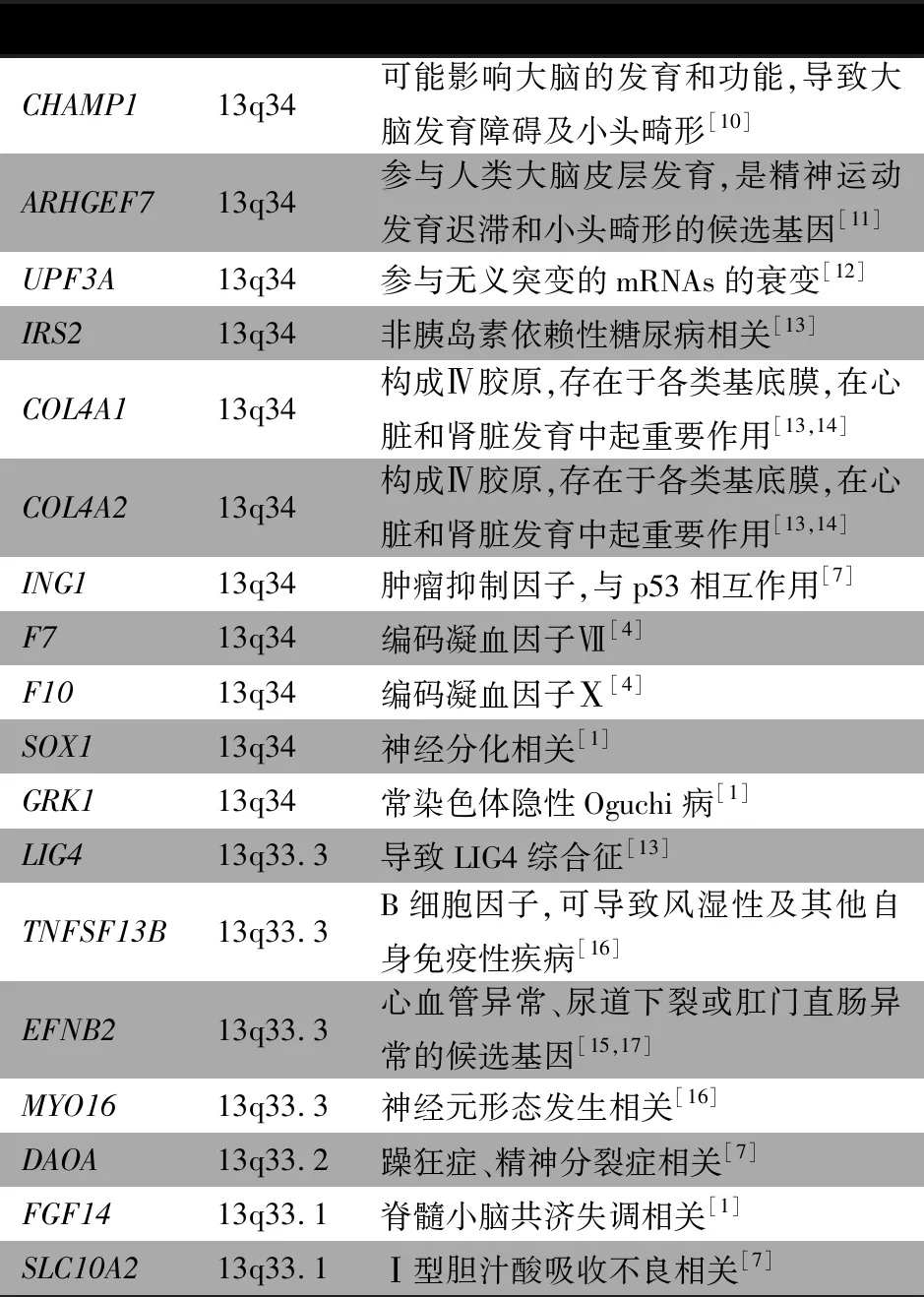
表1 13号染色体q33-q34缺失相关的基因定位及功能
本文患儿的染色体缺失范围为13q33.2-q34(Chr13:106213795-115107733),缺失区域包含27个基因。患儿在早期即出现血尿和蛋白尿,其后肾脏呈慢性病变,肾功能不全。推测本例患儿肾脏病变与13q33-q34缺失可能有关,因该区域包含多个发育相关基因,尤其是构成Ⅳ型胶原的同源基因COL4A1和COL4A2,这些基因的缺失或单倍体不足可导致肾脏病变,但具体发病机制尚未阐明。除了遗传因素,母亲怀孕期间服用致畸药物、酗酒等也可能影响胎儿肾脏发育。由于本文患儿未行全基因组检测,不能完全排除同时存在13q33-q34以外区域的基因存在致病变异的可能。此外,未对患儿父母做核型分析及基因芯片分析,无法明确患儿染色体缺失的来源。
回顾既往报道的13q33-q34缺失病例,有关肾脏病变的报道很少,其中1例2月龄男孩检查显示左肾缺如,但尿道无异常[8];1例7岁女孩尿常规显示镜下血尿,肾脏超声显示左肾较小,肾脏MR显示左肾发育不全,核素肾动态显像显示左肾肾小球滤过率明显低下,右肾肾小球滤过率正常[9]。本文患儿肾脏病变类型在既往13q33-q34缺失病例中未见报道。
本病目前尚无特效治疗手段,主要是对症支持治疗。由于涉及多系统异常,需要多学科评估及协助治疗,以最大程度地延长患儿寿命及改善患儿的生存质量。
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31 09:59:08
现代仪器与医疗(2022年1期)2022-04-19 13:52:04
新疆大学学报(自然科学版)(中英文)(2020年2期)2020-07-25 01:40:42
医学新知(2019年4期)2020-01-02 11:03:54
科学之谜(2019年3期)2019-03-28 10:29:44
科学之谜(2018年8期)2018-09-29 11:06:46
中国医药指南(2017年3期)2017-11-13 02:55:24
恋爱婚姻家庭·养生版(2016年9期)2016-09-07 11:25:01
中央民族大学学报(自然科学版)(2015年2期)2015-06-09 08:45:16
现代检验医学杂志(2015年1期)2015-02-06 01:59:23
