
高效液相色谱法测定氧化型染发产品中40种染发剂
2021-10-19 05:30邬国庆
色谱 2021年11期
左 雪, 邸 铮, 杜 勇, 杨 玲, 张 蓉, 邬国庆
(北京市药品检验所, 北京 102206)
随着居民生活水平的日益提升,我国染发类产品的市场需求也日趋增加。染发产品按作用机理可分为氧化型和非氧化型,其中氧化型为目前市场上销售、使用的主要产品类型。氧化型染发产品中的染发剂主要包括芳香胺类及酚类化合物等,相关研究表明,此类化合物具有不同程度的致敏性、致突变性及其他毒性作用[1-4],可引发皮肤过敏等不良反应[5-7],使该类产品的安全性备受关注。因此,针对氧化型染发产品中常见的染发剂,建立快速、准确的检测方法,为产品的研发与监管提供有效的技术手段,是十分必要的。
目前,包括液相色谱法[8-13]、液相色谱-质谱法[14-18]、气相色谱法[19]、气相色谱-质谱法[20,21]、离子色谱法[22]及毛细管电泳法[23]等在内的多种技术手段均已应用于染发剂的检测。其中,液相色谱法、液相色谱-质谱法应用最为广泛,但质谱仪器成本较高,且在分析复杂样品及成分含量未知的样品时,易造成离子源污染;液相色谱法适用于多种染发剂的分离分析,是一种比较理想的检验方法。查询、统计国家药品监督管理局2016~2018年注册的染发产品配方,并结合文献[24]报道,发现目前氧化型染发产品中添加的准用染发剂达四十余种。《化妆品安全技术规范》(2015年版,简称《规范》)[25]中提供的对苯二胺等32种组分的法定检验方法及严巍等[26]建立的32种禁限用染发剂的液相色谱测定方法,均可实现32种禁用、准用染发剂(其中包含24种准用染发剂)的检测,但需要同时建立3个液相色谱系统才能完成测定,仪器、色谱柱及试剂的消耗很大,且影响检验效率。近几年,多篇研究[11,27-29]报道了采用单一液相色谱体系同时测定多种染发剂的检测方法,方法较为简便、高效,但最多可检测不到30种染发剂。
本研究利用单一液相色谱系统建立了40种染发剂的定性、定量测定方法,其中包含36种准用染发剂。与《规范》[25]相比,该方法大大简化了液相色谱分析条件,并增加了1-羟乙基-4,5-二氨基吡唑硫酸盐、羟乙基对苯二胺硫酸盐、四氨基嘧啶硫酸盐、2,6-二羟乙基氨甲苯、2-氨基-6-氯-4-硝基苯酚、2-甲基-5-羟乙氨基苯酚、3-硝基对羟乙氨基酚、4-羟丙氨基-3-硝基苯酚、5-氨基-4-氯邻甲酚、5-氨基-6-氯-邻甲酚、HC黄2号、羟苯并吗啉和羟乙基-2-硝基对甲苯胺等13种常见准用染发剂,较大程度扩充了染发剂检测种类。与实验室前期建立的、同时测定33种染发剂的液相色谱方法[30]相比,该方法通过改变流动相种类、增加柱温变化等方式,进一步提升了各染发剂的分离效果,弥补了1-羟乙基-4,5-二氨基吡唑硫酸盐、四氨基嘧啶硫酸盐、2-氨基-3-羟基吡啶和对苯二胺等大极性组分分离度不佳的问题;该方法补充了《规范》[25]方法含有、而实验室前期建立方法中未包含的准用染发剂苯基甲基吡唑啉酮,同时还增加了《规范》[25]与前期方法中均未包含的2,6-二羟乙基氨甲苯等10种准用染发剂,实现了更多成分的有效分离与准确定量,提升了方法在产品实际检验工作中的全面性与针对性;方法通过提取溶剂浓度优化,进一步提高了样品提取率与检测结果准确性。该方法为氧化型染发产品的高效、经济、准确检测以及更全面监管提供了技术支持。
1 实验部分
1.1 仪器和试剂
1100型高效液相色谱仪配二极管阵列检测器(DAD),美国Agilent公司;CP225D型电子分析天平,德国Sartorius公司;KQ-500型超声波清洗器,昆山市超声仪器有限公司。
1,5-萘二酚等14种染发剂对照品购自北京曼哈格生物科技有限公司,1-萘酚等13种染发剂对照品购自德国Dr. Ehrenstorfer公司,2-氨基-6-氯-4-硝基苯酚等6种染发剂对照购自美国Sinco Pharmachem公司,2-甲基-5-羟乙氨基苯酚等3种染发剂对照品购自加拿大Toronto Research Chemicals公司,1-羟乙基-4,5-二氨基吡唑硫酸盐等3种染发剂对照品购自美国Ark Pharm公司,四氨基嘧啶硫酸盐购自北京百灵威科技有限公司。所有对照品纯度均不低于95%。40种染发剂对照品信息详见表1。

表 1 40种染发剂对照品的信息
乙腈(色谱纯)购自德国Merck公司;Milli-Q超纯水系统购自法国Millipore公司;乙酸铵(色谱纯)购自美国Fisher Chemical公司;无水乙醇(优级纯)购自现代东方(北京)科技发展有限公司;亚硫酸氢钠(分析纯)购自北京化学试剂公司。
样品均为市售氧化型染发产品,含染发膏和氧化乳两种剂型,取其中染发膏供含量测定。
1.2 溶液配制
含70%乙醇的亚硫酸氢钠水溶液:量取无水乙醇700 mL,置于1 000 mL量瓶中,加入2 g/L亚硫酸氢钠水溶液至刻度,充分振摇混匀,备用。
标准储备溶液:称取40种染发剂对照品各50 mg(精确至0.1 mg),置于同一50 mL棕色量瓶中,加入约40 mL含70%乙醇的亚硫酸氢钠水溶液,超声至充分溶解,再加入2 g/L亚硫酸氢钠水溶液至刻度,充分振摇混匀,即得混合标准储备溶液。
标准系列溶液:上述40种染发剂混合标准储备溶液用2g/L亚硫酸氢钠水溶液配制成系列标准溶液,质量浓度分别为5、10、50、100、250和500 mg/L。所得溶液于5 ℃条件下保存。
1.3 样品前处理
称取染发产品中染发膏0.5 g(精确至0.001 g),置于25 mL比色管中,加入含70%乙醇的亚硫酸氢钠水溶液至10 mL刻度,涡旋30~60 s至分散均匀,超声提取15 min,放冷后加入2 g/L亚硫酸氢钠水溶液至25 mL刻度,涡旋30 s,充分振摇混匀。经0.45 μm微孔滤膜过滤,续滤液待测。
1.4 色谱条件
色谱柱为Waters Atlantis®T3 MV Kit柱(250 mm×4.6 mm, 5 μm);流动相:A相为0.02 mol/L乙酸铵水溶液(含4%乙腈), B相为乙腈;流速为1.0 mL/min;进样量为5 μL。柱温及梯度洗脱程序见表2。同时设置2个波长进行检测,四氨基嘧啶硫酸盐、间苯二酚、1,5-萘二酚、2,7-萘二酚、HC黄2号、5-氨基-4-氯邻甲酚、羟乙基-2-硝基对甲苯胺、N-苯基-对苯二胺、1-萘酚等9种染色剂的检测波长为280 nm,其余染色剂的检测波长均为235 nm。样品盘温度设置为5 ℃。
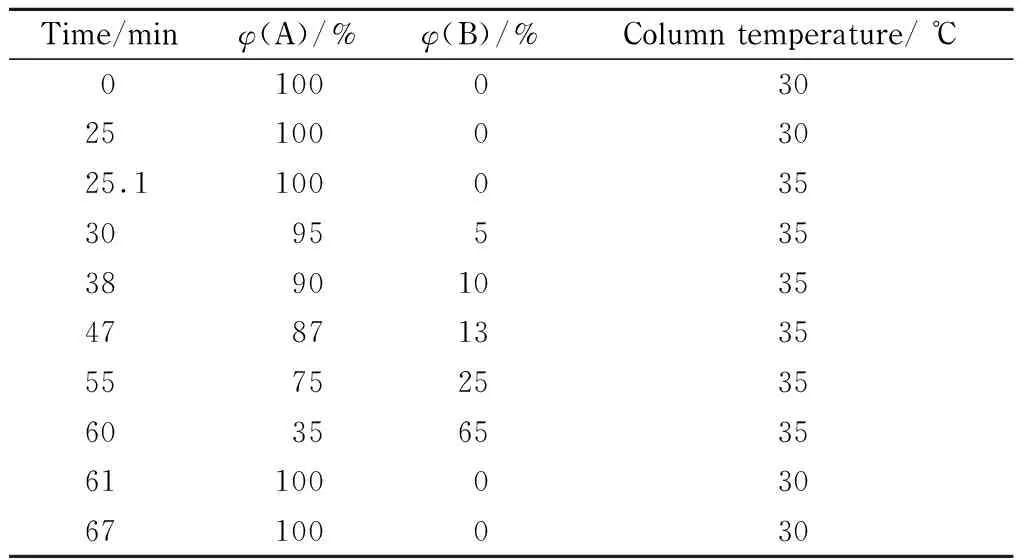
表 2 HPLC梯度洗脱程序
2 结果与讨论
2.1 流动相的选择
在实验室前期研究[30]中,选用磷酸盐缓冲体系(0.04 mol/L磷酸二氢钾和0.01 mol/L磷酸氢二钠,并含4%乙腈)和乙腈为流动相,通过梯度洗脱实现了33种染发剂的分离分析,但其中1-羟乙基-4,5-二氨基吡唑硫酸盐与四氨基嘧啶硫酸盐、2-氨基-3-羟基吡啶与对苯二胺、4-硝基邻苯二胺与4-氨基-3-硝基苯酚的分离效果有待提高。因此,本研究分别以上述磷酸盐缓冲体系-乙腈以及乙酸铵水溶液(含4%乙腈)-乙腈为流动相,比较两种体系对各种染发剂的分离效果。结果表明,乙酸铵体系(见图1a)对10 min内出峰染发剂的洗脱效果明显优于磷酸盐体系(见图1b),可有效解决极性较大染发剂的分离问题;两个体系(见图1c和1d)对10 min后出峰染发剂的洗脱效果相当。综上,选用乙酸铵体系作为流动相。在此基础上,比较不同浓度(0.01、0.02和0.03 mol/L)乙酸铵水溶液的分离效果,发现0.02 mol/L及0.03 mol/L乙酸铵水溶液的分离效果相当,均优于0.01 mol/L乙酸铵水溶液。从节约试剂及保护色谱柱的角度考虑,最终选择0.02 mol/L乙酸铵水溶液(含4%乙腈)和乙腈作为流动相。

图 1 采用不同流动相时染发剂混合标准溶液的色谱图Fig. 1 Chromatograms of dyes in the mixed standard solution using different mobile phases Mobile phases: for (a) and (c), (A) 0.02 mol/L ammonium acetate aqueous solution (containing 4% ACN) and (B) ACN; for (b) and (d), (A) 0.04 mol/L potassium dihydrogen phosphate and 0.01 mol/L disodium hydrogen phosphate (containing 4% ACN) and (B) ACN. Gradient elution procedure: 0-25 min, 0%B; 25-42 min, 0%B-14%B; 42-46 min, 14%B-40%B; 46-56 min, 40%B-60%B; 56-58 min, 60%B-0%B; 58-63 min, 0%B. Column temperature: 35 ℃; detection wavelength: 235 nm.
2.2 色谱柱及柱温的选择
继续使用前期研究[30]选定的Waters Atlantis®T3 MV Kit色谱柱(250 mm×4.6 mm, 5 μm)进行分离,并比较25、30和35 ℃柱温下的分离效果。结果表明,适当升温有助于色谱峰峰形的改善,可一定程度提高各染发剂的分离度。故进一步对30 ℃与35 ℃柱温条件进行比较,发现30 ℃柱温更利于25 min前出峰的染发剂的分离,而35 ℃更利于30 min后出峰的染发剂的分离。综上,考虑采用梯度变温的方式,25 min前柱温为30 ℃, 30 min后为35 ℃, 25~30 min色谱峰少,在此段时间内完成升温比较合适,故将此段时间作为变温过渡期。另外,为保证多次进样之间的重复性,在60 min后将温度降回初始温度30 ℃,并维持6 min,以实现系统平衡。经过多种尝试,最终确定1.4节描述的变温程序,确保绝大多数染发剂分离良好,且未见保留时间漂移。
2.3 检测波长的选择
采用DAD检测器在210~400 nm范围对40种染发剂进行全波长扫描,发现多数染发剂的最大吸收波长为220~245 nm,而235 nm下各染发剂均有较强的紫外吸收,认为该波长可以兼顾各成分的检测,因此首选235 nm为检测波长。但有9种染发剂存在特殊情况,具体如下:四氨基嘧啶硫酸盐、间苯二酚及N-苯基-对苯二胺在280 nm下吸收强于235 nm; 60 min后出峰的HC黄2号、5-氨基-4-氯邻甲酚、羟乙基-2-硝基对甲苯胺、N-苯基-对苯二胺及1-萘酚在235 nm下受到杂质峰干扰较严重;235 nm下,1,5-萘二酚、2,7-萘二酚及1-萘酚高浓度点响应过载。以上9种染发剂除了在低波长有最大吸收外,在275~310 nm有特征吸收,而280 nm可保证各成分的紫外吸收均较强,因此选择280 nm作为这9种染发剂的检测波长。利用1.4节色谱条件进行检测,得到40种染发剂在两种波长下的色谱图见图2。
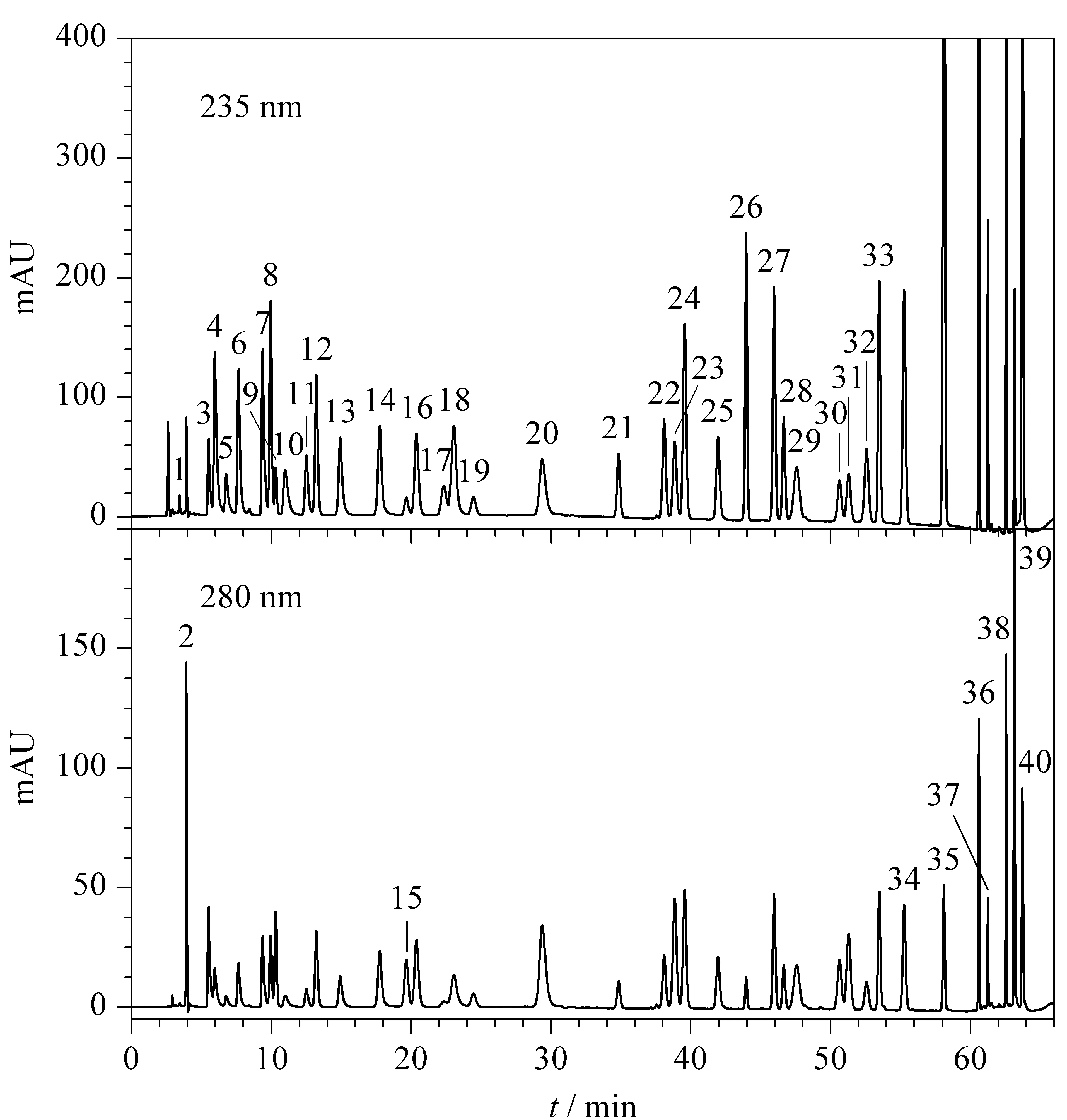
图 2 40种染发剂混合标准溶液在两种波长下的色谱图Fig. 2 Chromatograms of the 40 dyes in mixed standard solution at two wavelengthsPeak Nos. were the same as that in Table 1.
2.4 方法适用性
在确定流动相与色谱柱种类后,对梯度洗脱条件进行摸索与优化,最终确定1.4节下的色谱条件,利用此条件进行分析,40种染发剂可实现有效分离(见图2)。
同时发现,采用本文方法检测准用染发剂对甲基氨基苯酚硫酸盐时,其与N,N-双(2-羟乙基)-对苯二胺硫酸盐保留时间非常接近。实际检验过程中,若样品只含这2种染发剂中的1种,则可通过紫外光谱确定染发剂种类(见图3);若样品中同时含有上述2种染发剂,则可参考实验室前期建立的33种染发剂的测定方法[30],即采用25 ℃柱温,以磷酸盐缓冲液(0.04 mol/L磷酸二氢钾和0.01 mol/L磷酸氢二钠)-乙腈(96∶4, v/v)作为流动相进行等度洗脱,该方法可实现2种染发剂的有效分离与准确定量。

图 3 2种染发剂的紫外吸收光谱图Fig. 3 Ultraviolet absorption spectra of the two dyes a. p-methylaminophenol sulfate; b. N,N-bis(2-hydroxyethyl)-p-phenylenediamine sulfate.
在国家药品监督管理局网站上查询2016~2018年注册的1 000余种染发产品批件的配方信息。统计表明,3年注册的国产染发类产品中,未见对甲基氨基苯酚硫酸盐与N,N-双(2-羟乙基)-对苯二胺硫酸盐同时添加使用的情况;而仅7种进口产品同时添加了上述2种染发剂。可见,这2种染发剂同时添加使用的概率较低,故认为本方法适用于绝大多数氧化型染发产品中染发剂的测定。
2.5 提取溶剂的选择
依照《规范》[25]提供的样品前处理方法,取样品0.5 g,加入2 g/L亚硫酸氢钠水溶液-无水乙醇(1∶1, v/v)至10 mL,超声提取15 min后对所得样品溶液进行色谱分析,发现对苯二胺、2-氨基-3-羟基吡啶、羟乙基对苯二胺等极性较大的染发剂出现“溶剂效应”,即色谱峰“分叉”。而用2 g/L亚硫酸氢钠水溶液将上述样品溶液稀释至25 mL后,可有效解决此问题。因此,将样品前处理方法改为取样0.5 g,加入2 g/L亚硫酸氢钠水溶液和无水乙醇混合溶液至10 mL,超声提取后再加2 g/L亚硫酸氢钠水溶液至25 mL。
进一步比较含10%、30%、50%、70%及90%体积分数乙醇的亚硫酸氢钠水溶液对四氨基嘧啶硫酸盐、2-氨基-3-羟基吡啶、对苯二胺、甲苯-2,5-二胺硫酸盐、间氨基苯酚、间苯二酚、2-甲基间苯二酚、4-氨基-2-羟基甲苯、4-氨基-3-硝基苯酚、2,6-二羟乙基氨甲苯、4-氯间苯二酚、1-萘酚等12个使用频率较高且极性差异大的染发剂的提取效果。经对比,认为染发剂整体提取效果随乙醇体积分数的增大而提升。但发现,采用含90%乙醇的亚硫酸氢钠水溶液制备样品溶液时,即使加入亚硫酸氢钠水溶液稀释,对苯二胺等成分仍存在溶剂效应,色谱峰峰形不佳;采用含70%乙醇的亚硫酸氢钠水溶液提取时,整体效果仅次于90%组。综上,选定含70%乙醇的亚硫酸氢钠水溶液作为提取溶剂,并确定1.3节的样品前处理方法。
2.6 方法学考察
2.6.1标准曲线、检出限与定量限
在1.4节色谱条件下分析标准溶液,以染发剂的质量浓度(C, mg/L)为横坐标、峰面积(A)为纵坐标建立标准曲线,结果见表3。40种染发剂在各自范围内线性关系良好,相关系数(r)均大于0.999 8。
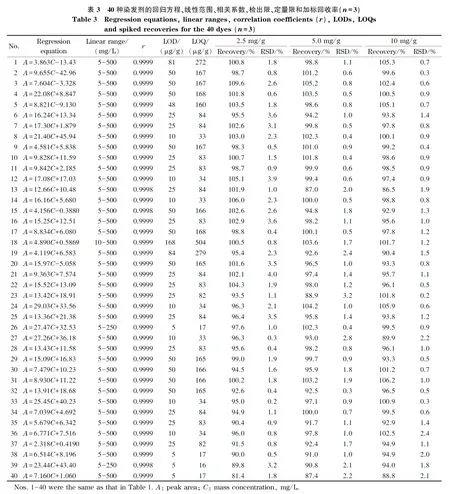
称取0.5 g(精确至0.001 g)空白基质样品(配方中不含本研究测定的40种染发剂),分别加入不同浓度的标准溶液,按1.3节方法进行前处理,分别以信噪比S/N为3和10时对应的含量作为检出限(LOD)与定量限(LOQ), 40种染发剂检出限为5~168 μg/g,定量限为16~504 μg/g,结果见表3。
2.6.2加标回收率
取基质空白样品,每份0.5 g(精确至0.001 g),分别加入标准储备溶液1.25、2.5和5.0 mL(相当于样品中染发剂含量分别为2.5、5.0和10 mg/g),每个水平制备平行样品3份,按1.3节方法进行前处理,在1.4节色谱条件下进行测定。计算各成分平均回收率及其RSD,结果见表3。40种染发剂的平均回收率为81.4%~109.6%, RSD值均小于5%。
2.6.3稳定性
取1.2节制备的100 mg/L标准溶液,按1.4节色谱条件分别于第0、4、8、12、16、20和24 h进行测定,计算各染发剂峰面积的RSD值。结果表明,40种染发剂在24 h内峰面积RSD值为0.2%~2.2%,说明稳定性良好。
2.7 实际样品测定
选取12批不同品牌、不同配方且添加染发剂种类较多的氧化型染发产品进行含量测定。12批样品中共检出24种准用染发剂,累计检出数量为66个,其在产品染发膏中的检出含量为0.01%~2.83%,全部符合《规范》[25]中的染发剂使用限值规定。所有样品均未检出对甲基氨基苯酚硫酸盐,1批样品中检出N,N-双(2-羟乙基)-对苯二胺硫酸盐,此染发剂具有单一紫外吸收光谱图谱,无对甲基氨基苯酚硫酸盐干扰,可利用本方法实现准确定量。
3 结论
本研究建立了高效液相色谱法测定氧化型染发类产品中40种染发剂,方法操作简便,准确性和稳定性良好,利用单一液相色谱系统进行分析,有效提高了实际应用中的检测效率;研究在《规范》检验方法基础上增加了13种准用染发剂,基本覆盖了氧化型染发产品配方中的常用染发剂。根据2016~2018年注册的染发产品配方统计结果,以及多个品牌产品的实际测定结果,认为本方法适用于绝大多数氧化型染发产品的检验检测。
猜你喜欢
中老年保健(2022年3期)2022-11-21
药学研究(2022年7期)2022-08-09
哈尔滨工业大学学报(2022年5期)2022-04-19
建材发展导向(2021年15期)2021-11-05
家庭医学(2021年9期)2021-09-28
家庭科学·新健康(2021年3期)2021-04-08
大众健康(2021年2期)2021-03-09
同济大学学报(自然科学版)(2019年8期)2019-08-07
自我保健(2019年5期)2019-08-02
同济大学学报(自然科学版)(2018年5期)2018-05-24
