
环糊精加速疏水缔合聚合物溶解机理研究
2021-10-18 12:20李玺罗平亚叶仲斌舒政赵文森肖秀婵
应用化工 2021年9期
李玺,罗平亚,叶仲斌,舒政,赵文森,肖秀婵
(1.成都工业学院 材料与环境工程学院,四川 成都 611730;2.西南石油大学 化学化工学院,四川 成都 610500;3.西南石油大学 油气藏地质及开发工程国家重点实验室,四川 成都 610500;4.海洋石油高效开发国家重点实验室,北京 100028)
疏水缔合聚合物是一种在传统的水溶性聚合物的主链上引入少量(通常摩尔含量<5%)疏水基团的新型水溶性聚合物[1],以其优异的流变性能受到油田化学的广泛关注。然而疏水基团间的相互作用强烈,导致疏水缔合聚合物溶解能力降低,不利于其在油田连续、大剂量使用,不能很好地满足不断发展的油田开发工程需要[2-3]。目前加速缔合聚合物溶解的方法都不可避免地造成缔合聚合物链段降解[4-8]。分析认为,疏水基团之间的缔合作用大幅延长了聚合物溶解过程中的松弛时间,而缔合聚合物溶解(浓稠体系逐渐稀释)过程实际上受松弛时间的控制[9]。如果能将缔合聚合物溶液的松弛时间降低,就能有效加速缔合聚合物的溶解。
环糊精(CD)是由α-1,4糖苷键连接吡喃葡萄糖单元而形成的环状多聚糖分子,其结构可视为截顶空心的圆锥体[10]。环糊精疏水的内腔与疏水基团有极强的亲和性,大量的文献报道了环糊精拆散缔合聚合物溶液中疏水基团形成的缔合结构的现象[10-13]。因此,本文利用环糊精对缔合聚合物疏水基团的亲和性,在缔合聚合物溶解时向溶剂中加入环糊精,以期这种化学方法能够加快疏水缔合聚合物的溶解速度。
1 实验部分
1.1 材料与仪器
疏水缔合聚合物AP-P4,由四川光亚聚合物化工有限公司提供;部分水解聚丙烯酰胺3630S(法国SNF),聚合物的主要理化性能指标见表1;α-CD、β-CD、γ-CD均由成都市科龙化工试剂厂提供;实验用水为模拟注入水,实验室自制,离子组成见表2。

表1 聚合物理化性能指标

表2 模拟注入水离子组成及含量
RW20Digital数显悬吊臂搅拌器;BROOK FIELDDV-Ⅲ黏度计;NikonSMZ1500光学显微镜;HAAKE MARS Ⅲ旋转流变仪;MS-TS电子天平(精度0.000 1 g)。
1.2 测定方法
1.2.1 流变测试 在7.34 s-1剪切速率下,用黏度计测定溶液的表观黏度;用旋转流变仪的锥板系统,在0.1~10 Hz内进行动态频率扫描,得到储能模量(G′)和耗能模量(G″)随频率的变化规律,测试温度25 ℃。
1.2.2 溶解时间测定 在25 ℃下,用模拟水配制5 000 mg/L的聚合物溶液。搅拌转速300 r/min,同时加入适量环糊精和过筛的聚合物干粉,每隔5 min测量溶液在剪切速率为7.34 s-1下的表观黏度,直至多次测定的溶液黏度值平稳。当黏度变化平稳(黏度变化率<5%)时认为聚合物基本溶解,此时间为聚合物的溶解时间。
1.2.3 溶胀时间测定 25 ℃条件下,在表面皿内加入一定量模拟水或者含有不同浓度β-CD的模拟水,放置在光学显微镜下。聚合物颗粒放入模拟水中,开始计时并启动自动照相系统进行连续摄像,对疏水聚合物溶胀过程进行观察。以聚合物颗粒完全透明作为实验终点,此时间为聚合物的溶胀时间。为避免粒径大小影响,尽量选取相近粒径(20~40目)的聚合物颗粒,多次重复实验,取平均值。
2 结果与讨论
2.1 两种类型聚合物的溶解
图1为AP-P4和3630S溶液黏度随时间变化曲线。

图1 AP-P4和3630S溶液黏度随时间变化曲线
由图1可知,聚合物黏度首先有一个缓慢增加的阶段,继而黏度迅速增大,再逐渐平稳或小幅波动(熟化阶段)。聚合物不能像小分子一样立即溶解,而溶剂分子较小,能迅速渗透进入聚合物。在溶解温度高于聚合物玻璃化温度以上时,聚合物的溶解通常分两步进行。首先溶剂分子渗入聚合物内部的自由体积和空隙中,使高分子体积膨胀——溶胀;其次,聚合物链段解缠结,均匀分散在溶剂中——溶解[9-10]。黏度缓慢增加阶段对应聚合物的溶胀过程,此时体系的黏度主要来自溶胀胶团之间的相互摩擦。随溶胀进行,胶团体积增加,体系黏度缓慢增加。随解缠结进行,聚合物链段舒展,流体力学半径扩大,体系黏度迅速增大,黏度迅速增加阶段对应聚合物的解缠结过程。在最后熟化阶段,解缠结继续进行,聚合物溶液从高浓度区域向低浓度区域迁移,最终形成均匀的聚合物溶液[14]。
虽然两种聚合物分子量接近,但AP-P4的溶解时间却比3630S长得多。而且AP-P4需要经过一个较长时间的黏度平稳期,而3630S的黏度增加较为迅速,平稳期不明显。这是因为缔合聚合物在溶解过程中需要克服的作用力与常规的HPAM溶解时需要克服的力不同。HPAM溶解时主要克服分子间作用力,缔合聚合物还需额外克服疏水基团之间的缔合作用。而疏水缔合能比分子间作用能和氢键的键能大得多,略小于比化学键的键能[1,6]。因此,分子量相近的缔合聚合物体系需要溶胀更长时间,使分子间距离增大到更大程度,才能开始溶解。宏观的表现就是溶解过程中溶液黏度增加更加缓慢,溶解时间比常规聚合物长得多。
2.2 环糊精对疏水缔合聚合物溶胀的影响
疏水缔合聚合物固体颗粒在溶胀过程中光学显微镜下形态见图2。聚合物固体颗粒不断吸水,形成半透明凝胶,未水化部分不断缩小。随溶胀过程继续,聚合物凝胶逐渐由半透明变得透明,未水化部分体积进一步缩小直至消失。

图2 AP-P4干粉颗粒溶胀过程(模拟注入水)
在含有不同浓度β-CD的溶液中,对20~40目AP-P4颗粒的溶胀时间进行多次测量,实验结果见表3。

表3 不同浓度β-CD溶液中AP-P4的溶胀时间
由表3可知,在模拟水含0.1%β-CD的模拟水和含0.5%β-CD的模拟水中,AP-P4颗粒平均溶胀时间分别为461,480,425 s。在相同溶剂中,AP-P4颗粒溶胀时间相对变化较大,且含有不同浓度β-CD的溶剂对溶胀时间的影响不明显。虽然AP-P4颗粒粒径接近,但是其溶胀时间仍然相差较大,这表明溶胀过程可能主要受到合成的聚合物胶体在后期干燥处理过程中形成的微小空隙和自由体积大小的影响[9]。
2.3 环糊精对疏水缔合聚合物溶解的影响
在含有不同摩尔比的环糊精/疏水基团(CD∶[H])的模拟水中,测定了缔合聚合物黏度随溶解时间的变化。为了便于不同实验之间相互比较,定义不同时间溶液表观黏度与最大表观黏度的比值为相对黏度。不同CD含量下溶液相对黏度与时间的关系曲线见图3~图5。
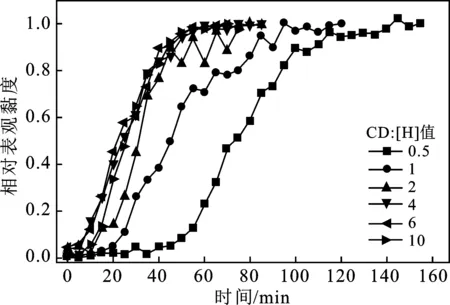
图3 不同α-CD∶[H]溶液中相对黏度与时间的关系曲线

图4 不同β-CD∶[H]溶液中相对黏度与时间的关系曲线

图5 不同γ-CD∶[H]溶液中相对黏度与时间的关系曲线
AP-P4在模拟水中溶解时的黏度缓慢增加时间持续约60 min,少量CD加入,缔合聚合物溶解时间就明显缩短。随CD∶[H]增加,AP-P4的溶解时间和黏度缓慢上升的时间都明显缩短。CD∶[H]继续增加,对溶解时间的缩短相对缓慢,且黏度缓慢增加的部分几乎观察不到,与常规HPAM表现出类似行为。在三种CD存在下,溶液相对黏度与溶解时间的关系曲线类似,聚合物溶解时间和黏度缓慢上升所耗时间都大幅度缩短。可能的原因是,未加入环糊精的条件下,缔合聚合物溶胀形成的凝胶层在分子间相互作用和强烈的缔合作用结合得比较紧密,难以通过搅拌从聚合物颗粒表面剥离[9]。同时,疏水缔合聚合物在溶液浓度超过临界缔合浓度之前分子内缔合占主导,溶液的黏度较低。即使有少量聚合物溶解,但由于溶液中的缔合聚合物浓度低于临界缔合浓度,分子内缔合占优,体系黏度也会一直保持较低值。加入环糊精后,屏蔽部分疏水缔合作用,凝胶层分子间的相互作用被削弱,更容易被物理搅拌所剥离。溶液中聚合物浓度增加更快,因而体系黏度增加更迅速。当CD∶[H]≥4,疏水基团之间缔合作用被屏蔽程度已经较高,溶液黏度缓慢增加段消失,表现出与常规HPAM类似的黏度-时间变化行为——加入聚合物干粉后溶液黏度直接迅速上升。
含有不同CD∶[H]的条件下,α-、β-、γ-CD与AP-P4溶解时间的关系见图6。

图6 CD∶[H]对AP-P4的溶解时间的影响
由图6可知,随CD∶[H]增加,溶解时间逐渐降低。液态水的结构由氢键结合的水分子簇组成,缔合聚合物溶解时疏水基团必然隔断周围水分子原来的氢键结构。氢键破坏,导致体系能量上升,在恒压下表现为体系的焓增加。而环糊精加入后,包合疏水基团,将疏水基团与溶剂的直接接触变为非直接接触,部分恢复了原来水分子的氢键结构,降低了溶解时的混合热(ΔHM)[15]。环糊精加量越大,对混合热降低得越多,ΔHM值愈小,自发溶解的倾向愈大[10,16]。
当CD∶[H]≤1时,随CD∶[H]增大,溶解时间大幅度下降。继续增大CD∶[H]比例,溶解时间缓慢减少。这是因为在CD∶[H]=1的模拟水中,缔合聚合物上大多数疏水基团已经被包合,溶解时间已经大大缩短。此后继续增大CD∶[H]比例,疏水基团被环糊精进一步包合的程度有限,因此缔合聚合物溶解时间缩短有限。到CD∶[H]=10时,α-CD、β-CD和γ-CD中的溶解时间分别为45,50,60 min,与3630S的溶解时间相当(65 min)。
对比三种环糊精在相同加量下对AP-P4溶解时间的影响,α-CD具有最强的加速溶解效果,β-CD其次,γ-CD最差。这可能跟环糊精疏水空腔的大小与疏水基团尺寸的匹配性相关。α-CD、β-CD和γ-CD 的疏水空腔直径分别为0.47~0.53 nm,0.60~0.65 nm和0.75~0.83 nm,而AP-P4的疏水烷基链的直径<0.49 nm[17],疏水基团跟α-CD的疏水空腔结合得比β-和γ-CD紧密。因此,α-CD对AP-P4加速溶解的效果更明显。
2.4 环糊精对疏水缔合聚合物溶液松弛时间的影响
通过动态频率扫描,得到β-CD对AP-P4溶液动态模量的影响,见图7。

图7 β-CD∶[H]对AP-P4溶液动态模量的影响
由图7可知,随CD∶[H]增加,弹性模量和黏性模量的频率依赖性增加。这表明聚合物溶液网络结构随CD∶[H]增加而逐渐削弱。根据瞬态网络理论[18-19],弹性模量与网络结构的活性连接点数量呈正比。弹性模量的下降,反映聚合物链段间活性链接数量的减少,表明环糊精包合疏水基团,屏蔽疏水缔合作用,导致缔合聚合物溶液结构破坏。
Gennes将橡胶状过渡到液体状的时间称为最终松弛时间(τt)[9]。τt也看作是高分子链的缠结被解开所需要的时间[20]。通常可以用低频下测量的最长松弛时间(TR)代表τt[21-22]。TR通过下式计算:
式中ω——角频率,rad/s;
G′——弹性模量,Pa;
G″——黏性模量,Pa。
将0.5%的AP-P4溶液的动态频率扫描数据通过式(1)进行计算,得到最长松弛时间(TR)随CD∶[H]变化情况,见图8。

图8 CD∶[H]对最长松弛时间/摩尔比(TR)的影响(0.5%)
由图8可知,随CD∶[H]增加,TR先迅速下降,表明解开链缠结所需时间随CD∶[H]增加而大幅减少。随CD∶[H]继续增加,最长松弛时间(TR)基本保持水平不再变化。CD∶[H]=20时,α-,β-,γ-CD分别将AP-P4最长松弛时间从最初的3.34 s缩短到0.25,0.36,0.50 s。虽然不能对溶解过程中所有浓度的溶胀胶团都进行相关的流变学研究,但根据瞬态网络理论,浓稠体系中聚合物松弛过程所遵循的规律应该是一致的,可以认为在溶解过程中,不同浓度溶胀胶团中高分子链的解缠结过程都随环糊精的加入而缩短。环糊精加速疏水缔合聚合物溶解是通过包合作用破坏疏水基团的缔合结构,缩短疏水缔合聚合物松弛时间而实现。此外,三种环糊精对最长松弛时间不同程度的影响还是应该归因于环糊精疏水空腔与疏水烷基链的空间匹配效应。
3 结论
疏水缔合聚合物溶解过程中,溶胀过程很短,而解缠结过程耗时较长。本文采用化学方法实现了疏水缔合聚合物的加速溶解,随环糊精加量增加,聚合物溶解时间迅速下降。环糊精对缔合聚合物的溶胀过程无明显影响,主要通过缩短缔合聚合物链段解缠结的过程而加速溶解。α-环糊精加速溶解效果最明显,β-环糊精次之,γ-环糊精效果最差,可能与环糊精空腔尺寸与疏水基团的匹配性有关。从热力学角度看,环糊精包合疏水基团,使疏水基团与溶剂的直接接触变为非直接接触,降低了缔合聚合物溶解的混合热,有助于提高聚合物自发溶解倾向。
猜你喜欢
小学阅读指南·低年级版(2022年5期)2022-05-09
当代水产(2021年10期)2022-01-12
云南化工(2021年11期)2022-01-12
中成药(2018年8期)2018-08-29
铜仁学院学报(2018年6期)2018-07-05
中成药(2018年4期)2018-04-26
衡阳师范学院学报(2016年3期)2016-07-10
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01
应用化工(2014年12期)2014-08-16
无机化学学报(2014年8期)2014-02-28
