
稀土改性多金属氧酸盐的合成、表征及催化燃油深度脱硫研究
2021-10-17 05:00赵庆辉宋应金徐昶儒刘日嘉
化学与粘合 2021年5期
王 斌,赵庆辉,刘 蕊,宋应金,綦 峥,徐昶儒,刘日嘉
(哈尔滨商业大学 药学院(药物工程技术研究中心),黑龙江 哈尔滨150076)
前 言
燃料是人类发展所必需的主要能源之一[1]。燃料的使用涉及到人类生活的各个方面,当燃料燃烧时会导致硫氧化物(SOx)的排放,会造成酸雨和大气污染。在机动车使用过程中,SOx的排放会引起汽车尾气催化转换器的的失活,影响其工作效率,从而导致有毒废气如碳氢化合物(HC)、一氧化碳(CO)、氮氧化物(NOx)的大量排放[2]。燃油的超深度脱硫可以从根本上降低环境污染对人类身体健康的危害。世界各国相继提出限制燃料油硫含量的法律法规,我国国Ⅴ标准以及欧Ⅴ标准中对汽油和柴油的硫含量限量不得超过10ppmw,2020年我国实行国Ⅵ标准,仍要求硫含量不高于10ppmw[3]。因此,生产超低硫燃料迫在眉睫,而从源头上降低燃油中硫化物含量就更显得尤为重要。
工业上常用加氢脱硫技术(HDS)脱除含硫化合物。然而,加氢脱硫反应条件苛刻,需要高温、高压,且由于噻吩类硫化物空间位阻的存在,该方法对噻吩及其衍生物等芳烃类含硫化合物的去除效果并不理想[4]。而相对温和的非加氢脱硫技术(NHDS)得到了广泛的研究,非加氢脱硫技术主要包括萃取脱硫[5]、吸附脱硫[6]、氧化脱硫[7]和生物脱硫[8]等,其中氧化脱硫技术因其反应所需条件温和、脱硫效率更高被认定为当今最具潜力的脱硫方式。在氧化脱硫过程中,硫化合物被氧化成相应的砜或亚砜,随后氧化产物可通过简单的萃取或吸附除去。氧化剂在氧化脱硫过程中起着至关重要的作用,常用的氧化剂包括过氧化氢(H2O2)和臭氧(O3)等[9~10]。
多金属氧酸盐(Polyoxometalates,POMs)是一种过渡金属氧簇类化合物,因其优异的氧化还原电位、高可控性、低温高活性、环境友好性和强酸性而被广泛应用于氧化脱硫反应中,在催化反应领域中十分重要[11]。刘日嘉等[12]制备了Keggin结构杂多酸盐Al0.5H1.5PW,并进行脱硫实验,实验结果表明:在一定条件下,反应到60min时,脱硫率可达99.3%。范铭晨等[13]用低温萃取-醚化法制备了多金属氧酸盐H3PMo6.1W5.9O40催化剂,结果表明:在最佳脱硫条件下,脱硫反应至7h,对模拟油品中BT的去除率可达100%。本研究通过分步反应,分步酸化合成了不同钒含量的硅钼钒前驱体,并通过稀土镧(La)、铈(Ce)、镨(Pr)、钕(Nd)、钐(Sm)、钆(Gd)进行改性,制备了多种稀土改性多金属氧酸盐,用于燃油超深度脱硫研究,为氧化脱硫在实际工业生产中的应用提供了一定的理论依据。
1 实验部分
1.1 试剂与仪器
硅酸钠(Na2SiO3·9H2O)、钼酸钠(Na2MoO4·2H2O)、乙腈(C2H3N)、正辛烷(C8H18)购于天津科密欧化学试剂有限公司;过氧化氢(H2O2)购于天津市大茂化学试剂厂;碳酸镧(La2(CO3)3)、硝酸铈(Ce(NO3)3)、硝酸镨(Pr(NO3)3)、硝酸钕(Nd(NO3)3)、硝酸钐(Sm(NO3)3)、硝酸钆(Gd(NO3)3)均购于上海阿拉丁生化科技股份有限公司,以上试剂均为分析纯。
WK-2F型微库仑综合分析仪(江苏江分电分析仪器有限公司);Controller B 180型马弗炉(德国Nabertherm公司);DH-101电热恒温鼓风干燥箱(天津市中环实验电炉有限公司);AL204分析天平(瑞士METTLER TOLEDO公司);FT-IR Spectrometer Frontier型傅里叶变换红外光谱仪(美国PerkinElmer公司)。
1.2 稀土改性多金属氧酸盐催化剂的制备
1.2.1 不同钒含量硅钼钒的制备
根据参考文献[14],制备不同钒含量的硅钼钒H4+xSiMo12-xVxO40·nH2O(x为1~4)
称取7.1g的硅酸钠(0.025mol)加入100mL蒸馏水溶解,记为溶液A;称取66.55g的钼酸钠(0.275mol)加入200mL蒸馏水进行溶解,标记为溶液B。称取2.925g偏钒酸铵(0.025mol)溶于80mL水中,标记为溶液C。将溶液A和B混合后置于油浴中,加热煮沸30min,在磁力搅拌下逐滴加入溶液C,保持油浴温度90℃下持续加热30min。继续搅拌,并用硫酸调节pH值在2.0~2.5范围内。冷却至室温,将溶液转移至分液漏斗中,加入足量乙醚,充分振荡后,加入一定量体积比为1∶1的硫酸溶液,继续充分振荡,静置,待溶液分层,将最下层溶液除乙醚、重结晶、干燥,得到硅钼钒H5SiMo11VO40,并将其标记为SiMoV1,不同钒含量硅钼钒H6SiMo10V2O40、H7SiMo9V3O40、H8SiMo8V4O40制备方法同上,并标记为SiMoV2、SiMoV3、SiMoV4备用。
1.2.2 稀土改性不同钒含量硅钼钒多金属氧酸盐的制备
采用重结晶法合成镧系稀土改性多金属氧酸盐[15~16]。称取5.34g SiMoV1(0.003mol)置于30mL体积比1∶1的乙醇溶液中。在75℃水浴剧烈搅拌,将镧系稀土化合物(0.0033mol)加入上述溶液中,混合溶液在常压蒸馏下搅拌蒸馏45min。停止加热,冷却至室温。抽滤,充分洗涤。随后将产物置于电热恒温鼓风干燥箱中烘干12h后,置于马弗炉中煅烧3h得到目标产物。将其记为LnSiMoVx(Ln代表La、Ce、Pr、Nd、Sm、Gd;x=1、2、3、4)备用。
2 结果与讨论
2.1 多金属氧酸盐结构表征
傅里叶红外光谱(FT-IR)是最常见杂多化合物的表征手段[17~18]。对参照1.2.2方法合成的稀土改性硅钼钒多金属盐酸盐LnSiMo10V2O40(Ln分别为La、Ce、Pr、Nd、Sm、Gd)进行红外光谱表征。Keggin结构的杂多酸化合物具有4个特征吸收峰,特征吸收峰在700~1100cm-1。如图1所示,谱图中1050~1100cm-1是Si-O键伸缩振动频率,900~1000cm-1是Mo-O键伸缩振动频率,850~900cm-1为M-O-M(不同组八面体氧桥)伸缩振动频率,750~800cm-1为M-O-M(同组八面体氧桥)伸缩振动频率,在1620cm-1左右出现的峰是H-O-H水分子弯曲振动。可以看到,稀土改性硅钼钒LnSiMo10V2O40(Ln分别为La、Ce、Pr、Nd、Sm、Gd)多金属酸盐催化剂,在以上指纹区均有对应的吸收峰出现,由此可以推断所合成的多金属氧酸盐催化剂具有Keggin型阴离子的典型特征结构。

图1 多金属氧酸盐红外光谱(a.LaH3SiMo10V2O40;b.CeH3SiMo10V2O40;c.PrH3SiMo10V2O40;d.NdH3SiMo10V2O40;e.SmH3SiMo10V2O40;f.GdH3SiMo10V2O40)Fig.1 The FT-IR spectra of the polyoxometalates
通过电感耦合等离子体发射光谱仪进行元素分析,用王水微波消解CeH3SiMo10V2O40,定容。配制标准溶液,上机测试分析。得到结果见表1所示,所合成的催化剂的钒含量稍稍偏高,可能是部分发生多个钒原子取代所造成的的影响[19],而其余各元素的实际含量与理论含量相近,可以证明所制得的催化剂原子组成与理论值相吻合。
2.2 稀土改性硅钼钒多金属氧酸盐催化性能考察
为了筛选不同原子配比及不同稀土改性的硅钼钒杂多酸盐的催化氧化脱硫性能,本实验以LaSiMoVx、CeSiMoVx、PrSiMoVx、NdSiMoVx、SmSiMoVx、GdSiMoVx(x=1、2、3、4)24种稀土改性硅钼钒杂多酸盐为催化剂,乙腈为萃取剂,在实验条件为:DBT作为典型硫化物,模拟油品初始硫含量为500ppmw,催化剂用量占模拟油品质量(m催化剂∶m模拟油品)的0.25%,氧硫物质的量比为20,催化剂与H2O2的预接触时间为1min,反应温度为60℃,反应时间固定为180min,脱硫率与时间的关系如图2所示。
由图2可以看出,LaSiMoVx、CeSiMoVx、PrSiMoVx、NdSiMoVx、SmSiMoVx、GdSiMoVx(x=1、2、3、4)24种稀土改性硅钼钒杂多酸盐催化剂均有较好的催化活性,待催化反应进行至180min时,对模拟油品中的DBT均有较高的去除效率,均达到超深度脱硫效果。由图2可以发现由铈改性的稀土多金属氧化物,相比于其他五种稀土多金属氧化物具有更佳的催化脱硫效率。由图中可以看出当钒原子取代数为2时,CeSiMoV2具有最强催化脱硫效率,在脱硫反应进行到30min时,模拟油品中DBT的脱除率已达100%。所以,在稀土改性硅钼钒多金属氧酸盐中,以Keggin结构的CeSiMoV2多金属氧酸盐作为代表性氧化脱硫催化剂,考察影响脱硫反应进程的各种条件,并对反应条件进行优化,进而提高CeSiMoV2的催化氧化脱硫效率。

图2 不同稀土改性的多金属氧酸盐的脱硫效率随时间的变化关系Fig.2 The changes of desulfurization efficiency of polyoxometalates modified with different rare earths with time

2.3 反应条件对模拟油品中DBT脱除效率的影响
本节实验考查了影响了Keggin结构多金属氧酸盐催化剂CeH3SiMo10V2O40催化脱硫效率的多种因素,如催化剂投加量、O/S物质的量比、催化剂与H2O2预接触时间、反应介质温度和初始硫含量,实验结果如图3所示。
由图3(a)可见,多金属氧酸盐CeSiMoV2对模拟油品中的硫化物均有较好的催化氧化脱除效果,在反应进行至180min时,对DBT的脱除率可达99.3%。当催化剂用量由0.125%增加到0.25%的过程中,催化剂的脱硫效率显著升高,这可能是因为催化剂与H2O2预接触的过程中生成了足够量的过氧阴离子{SiO4[MoO(μ-O2)(O2)]4}4-,提高了氧化剂的氧化活性,从而通过过氧阴离子的强氧化性促进了DBT的氧化反应进行[20]。当催化剂用量由0.25%增加至0.5%和1.0%时,从图中可以看到,随着催化剂用量的进一步增加,氧化脱硫体系的脱硫率增幅并不是十分显著。由图3(a)可见,当催化体系中不加入催化剂,仅仅通过萃取剂乙腈的萃取,待反应平衡时乙腈对硫化物的萃取率可接近50%,但随着萃取时间的延长,硫化物脱除率反而有些许降低,这可能是由于萃取反应平衡后,正辛烷相蒸发DBT量基本恒定所导致的。实验结果表明,CeSiMoV2具有极佳的催化氧化脱硫性能,综合经济因素和脱硫效果考虑,确定催化剂投加量为正辛烷质量的0.25%。
由图3(b)可见,多金属氧酸盐CeSiMoV2对模拟油品中的硫化物的脱除率与氧硫比有关(即H2O2的加入量),当氧硫比为0时,反应体系中仅有催化剂的存在。硫含量的降低是由乙腈的萃取所引起的。当萃取反应达到动态平衡后,硫含量不再变化,说明仅有催化剂存在时,是不能进行催化氧化脱硫反应的,只有催化剂与氧化剂二者相互反应,生成具有强氧化性的过氧阴离子,才能使氧化脱硫反应完成。由图3(b)可见,随着氧化剂量的增加,脱硫率也在逐渐升高,当氧硫比为2.5时,脱硫率为76.3%。当氧硫比为5时,脱硫率为83.5%,而当氧硫比为10和20时,在达到平衡时脱硫率可以达到98.0%,在一定浓度范围内,随着氧硫比的增加,催化氧化脱硫率也随之升高。说明在一定的氧硫比范围内,氧化剂的浓度越高,单位时间内多金属氧酸盐与H2O2预接触生成过氧活性中间体也越快,DBT的脱除效果越好。当催化反应达到平衡后,H2O2的增加并不会继续加快脱硫反应,这可能是由于副反应的发生影响了脱硫反应进程。综合能源消耗以及经济因素,将氧硫比定为10。
将催化剂与H2O2分别同时加入到氧化脱硫反应体系中,开始搅拌的时刻,记为0min。由图3(c)实验结果可以看到,当催化剂与氧化剂没有充分预接触时,催化剂催化氧化脱硫性能大大降低,这充分说明了催化剂与H2O2预接触的过程中生成足量过氧阴离子{SiO4[MoO(μ-O2)(O2)]4}4-,过氧阴离子的存在促进了氧化脱硫反应进程。从图中还可以发现,随着催化剂与H2O2的预接触时间的变化,脱硫效率显著升高,说明在一定时间范围内,催化剂与氧化剂预接触时间的增长会加快脱硫反应效率,促进反应向正向进行,但当催化剂与H2O2预接触时间升高至16min时,脱硫效率反而开始出现下降趋势,这可能是由于预接触时间过长导致过氧化氢在常态下分解所致[21]。从图3(c)中我们可以看出CeSiMoV2催化剂最优预接触时间为8min,在60min时先达到脱硫平衡,脱硫率能够达到95%以上。
由图3(d)可以看到,典型硫化物的脱除效率随着脱硫反应体系温度升高逐渐提高。当脱硫反应进行至120min时,各温度下的脱硫效率已基本达到平衡,且当反应体系温度为60℃和70℃时,催化剂的催化性能最佳。主要原因有:一是氧化脱硫反应为吸热反应,随着温度的升高更有利于脱硫正反应的进行;二是随着反应温度的升高,在脱硫反应体系中催化剂与H2O2的有效碰撞几率升高,随之生成更多更具活性过氧活性中间体,从而提高氧化脱硫效率。由图3(d)可知,在反应体系温度为60℃和70℃下,DBT的脱除率并无显著差异,甚至在反应体系60℃下的脱硫效率更高一些,主要原因为:一是氧化脱硫体系中对DBT的氧化脱除反应符合一级反应动力学,更高的反应温度只能缩短反应时间,由于反应速率的限制脱硫效率并不会显著影响脱硫率。二是更高的体系温度,会增加正辛烷的挥发,进而造成单位体积内DBT含量的升高。因此,综合考虑脱硫效果和能耗的影响,将氧化脱硫体系的温度确定为60℃。
由图3(e)我们可以得出,在90min达到脱硫平衡时,催化剂对于500ppmw以下模拟油品脱除效率较好,脱硫率可达到97%以上。而500ppmw模拟油品的脱硫率接近于100%。达到了深度脱硫的效果,说明在一定范围内,随着模拟油品硫含量的升高,脱硫率也是随之升高。可能是由于浓度升高,催化剂与反应物之间的接触机会增多,所发生的有效碰撞也随之增加。而800ppmw的脱硫率反而下降,可能是催化剂与H2O2预接触生成的过氧阴离子,不足以完成对800ppmw硫化物的脱除,从而使脱硫率有所降低。综合分析,将初始硫含量定为500ppmw。
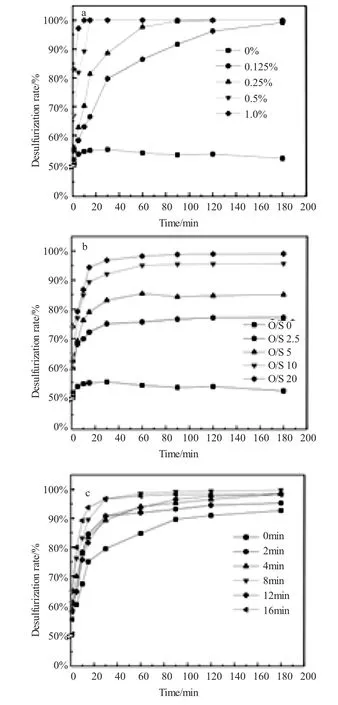

图3 反应条件对模拟油品中DBT脱除效率的影响(a:催化剂用量、b:O/S物质的量比、c:催化剂与氧化剂H2O2预接触时间、d:反应介质温度、e:初始硫含量)Fig.3 The effect of reaction conditions on the simulated oil DBT removal(a:catalyst dosage,b:O/S molar ratio,c:pre-contact time between the catalyst and the oxidant H2O2,d:reaction temperature and e:initial sulfur content.)
2.4 催化剂对不同噻吩类硫化物脱除效果的影响
实验结果如表2所示,以多金属氧酸盐CeSiMoV2作为催化剂时,在最佳催化条件下,当脱硫反应至180min时,4种噻吩类含硫化合物TH、BT、DBT、4,6-DMDBT的最终脱硫率分别为64.44%、78.63%、100%、89.53%。对不同噻吩类硫化物的氧化脱除顺序为DBT>4,6-DMDBT>BT>TH。由表2所示的各噻吩类硫化物的电子云密度与氧化脱硫效率可以看出,不同硫化物的氧化脱硫反应活性与硫原子电子云密度呈正相关,TH的电子云密度最低,反应活性最弱,而4,6-DMDBT在其分子结构上存在两个甲基,甲基的存在产生空间位阻效应,它的电子云密度高于DBT,由于其空间位阻的影响占主导因素,导致其脱硫效率活性不及DBT。

表2 多金属氧酸盐CeSiMoV2对不同噻吩类硫化物的脱除效果Table 2 The removal of different thiophene sulfides by the polyoxometalate CeSiMoV2
2.4 真实燃油催化氧化脱硫性能研究
本节实验以稳定汽油和催化柴油作为研究对象,在最佳实验条件下,待脱硫反应至180min时,稳定汽油和催化柴油中剩余硫含量分别为102.53ppmw和340.98ppmw(见表3)。脱硫效果不及模拟油品的主要原因为:一是真实燃油成分复杂,含有多种不易脱除的硫化物。二是真实燃油炼制过程中会加入一些添加剂,添加剂的加入会引起部分催化剂失活,从而导致催化剂的活性降低,进而影响催化剂的催化效果。为解决以上问题,我们将脱硫完成后的真实油品进行连续分级萃取。对稳定汽油进行连续分级萃取,萃取四次后硫含量降至7.48ppmw。对催化柴油进行连续分级萃取,萃取6次后硫含量降至7.56ppmw,均符合我国现行的国Ⅵ标准。

表3 脱硫后稳定汽油与催化柴油连续萃取硫含量变化Table 3 The changes of sulfur content in continuous extraction of stabilized gasoline and catalytic diesel after the desulfurization
2.5 催化剂重复利用
催化剂CeSiMoV2回收后进行催化氧化脱硫实验,经过5次重复利用后,脱硫率分别为99.52%、99.05%、98.48%、97.85%和97.33%。催化性能未见明显降低,实验结果表明多金属氧酸盐CeSiMoV2作为催化剂重复利用效果良好。
3 结 论
本文通过分步反应,分步酸化合成了不同钒含量的硅钼钒前驱体,并通过稀土镧(La)、铈(Ce)、镨(Pr)、钕(Nd)、钐(Sm)、钆(Gd)进行改性,制备了多种稀土改性多金属氧酸盐,通过对催化剂的催化性能进行筛选,确定最优催化剂为CeH3SiMo10V2O40。在最佳催化条件(催化剂用量占模拟油品质量的0.25%、氧硫比为10、催化剂与H2O2预接触时间为8min、反应温度60℃、初始硫含量500ppmw)下,当反应达到平衡时,模拟油品中硫化物已完全脱除,达到超深度脱硫目的。并将CeH3SiMo10V2O40用于真实燃油脱硫实验中,也得到了较好的催化脱硫效果。结果表明,多金属氧酸CeH3SiMo10V2O40对模拟油品及真实燃油均具有较高的催化脱硫效果,且重复使用效果良好,为多金属氧酸盐催化剂催化氧化有机硫化物的实际应用提供了有效的理论支撑。
猜你喜欢
金属热处理(2022年10期)2022-10-25
昆明理工大学学报(自然科学版)(2022年4期)2022-09-07
加油站服务指南(2022年6期)2022-07-28
昆钢科技(2022年2期)2022-07-08
中国农业科学(2022年12期)2022-06-28
船舶经济贸易(2022年5期)2022-06-02
电工材料(2022年2期)2022-04-26
化工技术与开发(2021年10期)2021-10-27
当代水产(2021年3期)2021-07-20
科学与财富(2021年35期)2021-05-10
