
视网膜色素变性和视锥-视杆细胞营养不良患者基因突变频谱分析
2021-10-13 12:19任英华盛迅伦容维宁
国际眼科杂志 2021年10期
任英华,盛迅伦,贾 沁,容维宁,张 爽
•KEYWORDS:autosomal recessive inheritance; retinitis pigmentosa; cone-rod dystrophy; gene detection; mutation analysis
0引言
遗传性视网膜变性疾病(hereditary retinal dystrophies, HRD)是临床上最常见的致盲性遗传性视网膜变性疾病[1],主要病因为遗传性基因突变,具有高度的临床和遗传异质性。其中常染色体隐形遗传视网膜色素变性(autosomal recessive retinitis pigmentosa, ARRP)和常染色体隐形遗传视锥-视杆细胞营养不良(cone-rod dystrophy, CORD)两种疾病的表型之间具有一定程度的相似性和交叉性,致病基因又有重叠性,从遗传学及临床表型探索疾病的特点有利于发现疾病规律,进一步寻找共同治疗靶点。视网膜色素变性(retinitis pigmentosa, RP)是由于光感受器(视杆细胞和视锥细胞)或视网膜色素上皮的结构或功能异常相关的一类遗传性进行性致盲眼病,其发病机制尚未完全明确,在由单一基因所致盲的遗传性眼病中RP发病率占首位。世界范围内的发病率为1/4000[2],影响全世界约250万人[3]。CORD以视锥细胞受损为主,伴不同程度的视杆细胞损伤。两者均为高度临床异质性和遗传异质性眼病,许多基因缺陷均可导致上述两种疾病的发生,目前不能通过基因检测结果直接进行准确诊断,限制了本病基因诊断在临床的广泛应用。本研究应用目标序列捕获结合二代测序技术对ARRP和CORD患者进行基因突变频谱检测分析,同时结合临床表型分析,初步探讨ARRP和CORD的诊断和鉴别诊断。
1对象和方法
1.1对象选择2016-09/2020-02在宁夏眼科医院就诊的35例ARRP患者和18例CORD患者纳入研究,采集先证者及家系成员病史资料,所有患者及其家庭成员均接受详细的眼部检查,包括裂隙灯显微镜、间接检眼镜、裸眼视力、最佳矫正视力、视野、彩色眼底照相、频域光相干断层扫描(optical coherence tomography, OCT)、荧光素眼底血管造影及视网膜电图(electroretinogram, ERG)等检查。ARRP纳入标准:(1)符合ARRP的诊断标准:1)首发夜盲史;2)视力逐渐下降;3)眼底表现为视盘色蜡黄或变淡,视网膜血管变细,周边视网膜可见骨细胞样色素沉着,晚期视野呈向心性缩窄;4)暗适应检查早期视杆细胞功能降低,视锥细胞功能正常,随着病情的进展,视锥视杆细胞功能均下降甚至丧失;(2)通过系谱分析确定为RP患者。ARRP排除标准:通过全身检查排除综合征性RP患者、非典型RP患者。常染色体隐性遗传CORD纳入标准:(1)符合CORD的诊断标准:1)首发视力逐渐下降;2)视野出现中心暗点;3)全视野ERG出现视锥视杆细胞功能均下降甚至丧失,并且视锥细胞功能和视杆细胞功能相同或受损更严重;4)多数患者眼底检查和黄斑OCT检查显示黄斑萎缩;(2)通过系谱分析确定为CORD患者。常染色体隐性遗传CORD排除标准:通过全身检查及基因检测排除误诊为CORD患者。本研究遵循赫尔辛基宣言,并经宁夏回族自治区人民医院宁夏眼科医院伦理委员会审核通过批文号,所有研究对象和未成年患者监护人均签署知情同意书。
1.2方法
1.2.1DNA提取及测序分析从受检者外周血中提取DNA。应用的目标序列捕获芯片包括232个由RetNet网站(https://sph.uth.edu/RETNET)所公布的HRD已知致病基因,针对已知的致病基因及突变位点定制特异性的寡核酸苷酸探针,通过Agilent SureSelect外显子靶向序列富集系统对目标基因组区域进行液相捕获,高通量测序仪(Illumina HiSeqTM2000)进行测序。将高通量二代测序结果应用Burrows-Wheeler Aligner软件同University of California Santa Cruz(UCSC)人类基因组对照序列hg19完成比对,并运用Genome Analysis Tool Kit工具对获得的测序结果进行校准及完善。将检测获得的DNA序列变异信息与单核苷酸多态性数据库进行比对过滤,应用ANNOVAR对剩余的突变基因进行蛋白质的改变预测,剔除同义突变,利用专业版人类基因突变数据库进行基因位点检索获得候选致病突变位点。对可疑致病变异进行Sanger验证及家系共分离分析。
1.2.2数据分析依据美国医学遗传学与基因组学学会(American college of medical genetics and genomics, ACMG),2015年发布的《序列变异解读标准和指南》对新发变异进行基因变异致病性评估。已报道变异分类标准参考文献方法[4],采用GERP++软件和UCSC(http://genome.ucsc.edu/)在线工具对突变位点的氨基酸进行保守性分析。在1000Genome(http://browser.1000genomes.org/index.html)、EVS(http://evs.gs.washington.edu/EVS/)和ExAC(http://exac.broadinstitute.org/)数据库中查看变异在正常人群中的等位基因频率,最小等位基因频率小于0.005作为排除良性变异的标准。选用Polyphen-2(http://genetics.bwh.harvard.edu/pph2)、SIFT(http://sift.jcvi.org)、PROVEAN(http://provean.jcvi.org/index.php)和MutationTaster(http://www.Mutationtaster.org)进行致病性预测。用Human Splicing Finder和MaxEntScan(http://www.umd.be/HSF/)预测变异对pre-mRNA正确剪切的影响。使用HOPE(https://www3.cmbi.umcn.nl/hope)分析变异对于编码蛋白结构的影响。
2结果
有些遗传性眼病家系中除先证者外,家庭成员中暂找不到其他患者,无法确定其遗传方式,既往称为散发型。但是随着近年来二代测序技术的广泛应用,许多对散发遗传性眼病的研究表明这些病例大部分属于常染色体隐性遗传。本研究的35例ARRP患者中,首发症状为夜视力下降者26例,不同程度进行性视力下降者为5例,健康查体发现为4例,首发症状年龄为6~49岁,最佳矫正视力范围为0.1~2.0。本研究的18例CORD患者中,首发症状为进行性视力下降,首发症状年龄为12~36岁,最佳矫正视力范围为指数/30cm~0.3。
本研究中对35例ARRP患者和18例CORD患者进行基因检测分析,其中表1提示:在35例ARRP患者中,检测到突变基因共有16个,分别是ABCA4、C2orf71、C8orf37、CLRN1、EYS、CLN3、CRB1、CNGA1、KIAA1549、MERTK、NRL、PROM1、RDH12、RHO、RP1和TULP1,其中以RP1基因突变率最高,占14%(5/35),其次为以ABCA4、CRB1和EYS基因,均占11%(4/35);表2提示:18例CORD患者中,共检测到突变基因10个,分别是ABCA4、ALMS1、CLN3、CRB1、NRL、PROM1、RPE65、USH2A、GUCY2D、CDH23,其中以ABCA4基因突变率最高,占28%(5/18),其次为以ALMS1、PROM1、RPE65、USH2A基因,均占11%(2/18);在ARRP和CORD患者中,共同致病基因有ABCA4、CLN3、CRB1、PROM1、NRL共5个,占42%(22/53)。

表1 ARRP患者中突变基因位点
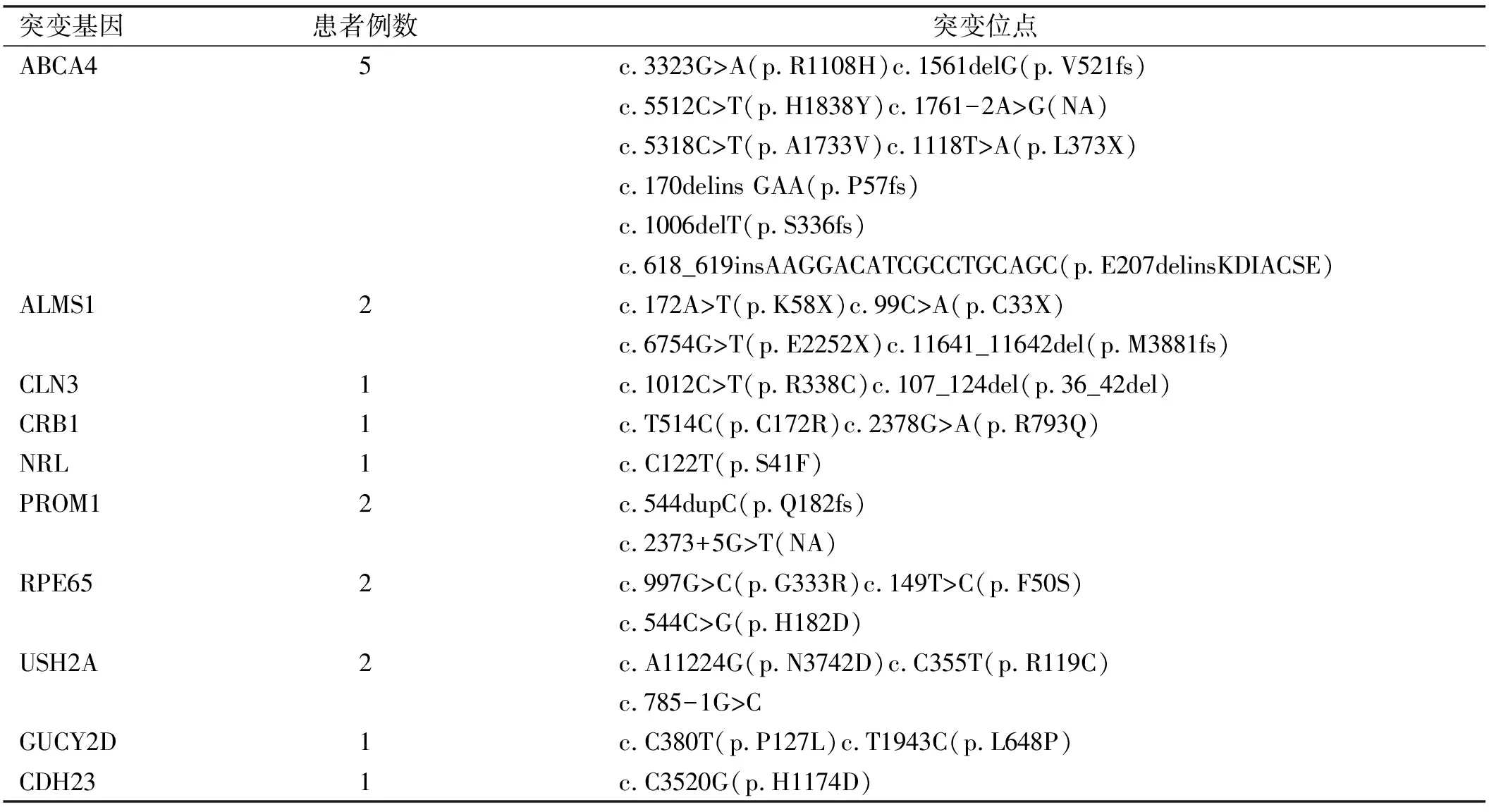
表2 常染色体隐性遗传CORD患者中突变基因位点
3讨论
在HRD的研究中,致病基因临床表型存在很大的异质性。研究HRD表型异质性有利于丰富疾病表型谱特征,促进HRD的精准诊断和个体化治疗。本研究应用目标序列捕获结合第二代测序技术对患者进行突变基因筛查,筛查技术快速而准确。在ARRP和CORD两种疾病中,ARRP共筛查出16个致病基因,CORD共筛出10个致病基因,覆盖范围为常见致病基因,表现为高度的遗传异质性和临床异质性,其中有5个共同致病基因,分别为ABCA4、PROM1、CLN3、CRB1、NRL。ABCA4的检出频率最高,其次为PROM1。根据RetNet网站公布的数据(RetNet:https://sph.uth.edu/Retnet/sum-dis.htm)显示,ABCA4、C8orf37、CERKL、PROM1共4个基因既可以导致ARRP又可导致CORD。由此可见,以上ARRP和CORD两种疾病的致病基因具有重叠性,本研究结果中检测出5个共同致病基因也证实了这一结果。由于ARRP和CORD两种疾病无论在表型上还是在疾病进展严重程度上均表现一定程度的相似性和交叉性,在不同年龄阶段和疾病发展的不同时期具有明显的临床异质性,在致病基因上也存在一定重叠,两种疾病表型主要区别在于首发临床症状,ARRP首发症状为夜盲,该症状可持续多年后出现视力下降,此时ERG检查表现视锥视杆细胞均重度下降。本研究中患者首诊原因主要为夜视力下降,最佳矫正视力范围0.1~2.0,视盘颜色明显变淡或蜡黄,视网膜血管变细,后极部及周边散在骨细胞样色素颗粒沉着。而CORD首发症状主要为视力下降,主要为视锥细胞功能障碍伴较晚的视杆细胞受累,黄斑区视网膜外层光感受器受损,随年龄增长范围扩大,可累及周边部甚至全视网膜,ERG检查视锥细胞反应可呈熄灭型或明显下降。
ABCA4基因突变能够引起Stargardt病(STGD)、COD/CORD、ARRP或部分类型的年龄相关性黄斑变性等临床表型,且患者的发病年龄、发病特点也各不相同,ABCA4基因突变人群发生率是4.5%~10%[5]。ABCA4基因定位于1p22.1,主要编码ABCA4蛋白,ABCA4蛋白是感光细胞中重要的功能性蛋白。功能正常的ABCA4蛋白能够将视循环视黃醛代谢的中间产物N-视黄基磷脂酰乙醇胺转运,帮助其自发分解为全反式视黄醛,进入视循环。当ABCA4蛋白功能异常时,N-视黄基磷脂酰乙醇胺聚集,并随感光细胞脱落至RPE中,经进一步反应生成N-亚视黄基-N-视黄基乙醇胺(A2E),A2E是脂褐质的主要基团,具有细胞毒性,会导致RPE细胞凋亡[6],进而破坏视网膜色素上皮的结构和功能,导致疾病发生和发展。一般常染色体隐性遗传发病早,病情进展快。视功能损害较为严重。王晓光等[7]研究表明,ABCA4基因突变出现在CORD患者及其同胞妹妹基因检测中,二者虽致病基因一致,但眼底表现存在差异,因此,仅通过基因检测难以做出准确诊断,需与临床表型相结合分析才能确定诊断。本研究中,ABCA4重叠出现的频次最高,且是由不同突变位点导致不同疾病的发生,临床表现也出现多样化。
PROM1基因定位于4p15.32,在视网膜中的确切生理作用尚不清楚,PROM1相关的临床表型主要包括STGD,CORD等。PROM1蛋白定位于光感受器外段[8-9]。有研究发现PROM1-/-小鼠表现出Rdh12和ABCA4的显著下调,并推测PROM1缺乏的下游事件是A2E/脂褐素的累积,并加重ABCA4基因突变的效应[10]。研究发现PROM1相关的形态表型均与CORD相关[11]。本研究中ARRP家系的PROM1基因突变位点为c.27_28del(p.L9fs)c.2309C>A(p.P770H)和c.2788-52C>A(NA),成员尚未见锥杆细胞营养不良,可能与疾病发展阶段相关,需要长期随访。CORD家系的PROM1基因突变位点为c.544dupC(p.Q182fs)和c.2373+5G>T(NA)。
CLN3基因首先被报道为神经元蜡样脂褐质沉积症的致病基因,基因定位于染色体16p12.1~p11.2[12],编码的蛋白是一个完整的膜蛋白,眼内CLN3主要定位于视网膜感光细胞的内段、视网膜内部细胞和Müller细胞中,与高尔基体和突触蛋白的运输相关[13],为凋亡抑制基因。其突变常与神经退行性疾病相关,眼病患者表现为CORD,其视杆细胞功能降低明显,而视锥细胞功能多变[13]。最早关于CLN3的研究是Batten病,这是一种感光细胞及神经细胞过度凋亡引起的常染色体隐性遗传的神经退行性疾病,病理表现为脑神经元细胞的凋亡及内源性神经酰胺水平升高[14],患者的CLN3有近1kb的基因缺失,临床表现为精神、运动系统退行性疾病迅速进展,癫痫发作、发育不良、小头畸形,共济失调和感光细胞退化而导致的视力丧失。2014年,Wang等[15]报导CLN3可引起ARRP和CORD,但未见全身症状。本研究中ARRP患者CLN3基因突变位点为c.416G>A(p.G154D),而CORD患者基因突变位点为c.1012C>T(p.R338C)c.107_124del(p.36_42del),未发现神经系统异常,伴随疾病的发展,是否会出现神经系统异常需要长期随访观察。
CRB1基因定位于1q31.3,要在光感受器内段以及大脑中表达,主要参与光感受器形态形成,突变可能抑制视网膜的发育,导致光感受器信号传导的丢失[16]。CRB1突变的临床表型主要有RP和Leber先天性黑朦[17],本研究在CORD中也发现2例CRB1基因突变,突变位点分别为c.T514C(p.C172R)和c.2378G>A(p.R793Q)。
NRL基因定位于14q11.2,编码合成碱性亮氨酸拉链蛋白,属于DNA结合蛋白的亮氨酸拉链家族。对视杆细胞的发育和分化具有重要作用。表达于成熟视网膜中。在模仿人类视网膜的发育研究中发现NRL敲除会导致视杆细胞的完全缺失[18],并且NRL也是RP基因治疗的靶点之一[19]。
综上所述,目标区域测序的基因诊断方法是检测HRD的强大工具,结合临床表型才能更准确的做出临床诊断。在ARRP和CORD两种疾病中,存在表型异质性和致病基因的重叠,同一基因不同位点突变可以导致不同疾病或同一疾病但不同临床表型。因此,对ARRP和CORD的诊断需要依据基因检测结果和更多更深入的临床研究。遗传性眼病表型复杂,明确基因型和临床表型关系有利于辅助基因治疗的研究。
猜你喜欢
中华医学图书情报杂志(2022年1期)2022-11-18
中国现代医生(2022年21期)2022-08-22
农村科学实验(2022年2期)2022-03-12
三农资讯半月报(2020年2期)2020-03-09
少儿科学周刊·儿童版(2019年6期)2019-08-24
科学之谜(2018年9期)2018-12-17
小学生导刊(2018年13期)2018-06-29
中学生理科应试(2017年6期)2017-09-27
方圆(2016年8期)2016-05-04
中学生天地(A版)(2009年12期)2009-05-28
