
Comparison of fungal community composition within different intestinal segments of tilapia and bighead carp*
2021-10-12 05:01:38LeiZHOUYaoquanHANDapengWANGYusenLIXiandeHUANGAnyouHE
Lei ZHOU ,Yaoquan HAN ,Dapeng WANG ,Yusen LI ,Xiande HUANG ,,Anyou HE
1 Guangxi Key Laboratory of Aquatic Genetic Breeding and Healthy Aquaculture, Guangxi Academy of Fishery Sciences, Nanning 530021, China
2 Joint Laboratory of Guangdong Province and Hong Kong Region on Marine Bioresource Conservation and Exploitation,College of Marine Sciences, South China Agricultural University, Guangzhou 510642, China
Abstract Although intestinal fungi play important roles in host health and disease,the composition and diversity of fungal communities remain poorly reported in fish.In this study,fungi in the fore-,mid-,and hindintestine of tilapia (Oreochromis mossambicus) and bighead carp (Aristichthys nobilis) from Hongchaojiang Reservoir in Guangxi,China were investigated by ITS sequencing.Based on this,we obtained 1 763 478 high-quality tags,which clustered into 1 089 operational taxonomic units (OTUs).In total,404 OTUs were annotated,of which 310,68,and 26 belonged to Ascomycota,Basidiomycota,and other,respectively.Results show significant differences in the community composition of intestinal fungi between tilapia and bighead carp but not within their different intestinal segments.Furthermore,154 of the 404 annotated OTUs were considered reliable and were classified into three trophic modes and nine guilds.The three trophic modes consisted of 108 OTUs of saprotrophic fungi,41 OTUs of pathotrophic fungi,and five OTUs of symbiotrophic fungi.The top three most abundant OTUs overall (i.e.,Otu000002,Scopulariopsis acremonium;Otu000018,Alternaria palandui;Otu000034,Aureobasidium pullulans) showed lower abundance in the hind-intestinal segments of bighead carp,suggesting uneven distribution of these fungi in this species.In addition,saprotrophic and pathotrophic fungi were markedly decreased in the hindintestine.It is indicated that the fungal community was not only related to host species specificity but also to the respective physiological functions of different intestinal segments.These findings provide valuable information on the composition,structure,and potential function of the intestinal fungi community associated with different intestinal segments in tilapia and bighead carp under natural conditions,thus highlighting the importance of fungi as an integral part of the intestinal microbiota in maintaining host health.
Keyword: fungi;intestinal microbiome;tilapia;bighead carp;Internal Transcribed Spacer (ITS)sequencing
1 INTRODUCTION
Host-associated microorganisms are vital for host species growth and survival (Eichmiller et al.,2016).In particular,the vertebrate intestinal tract is a complex ecosystem that provides optimal habitat for dynamic microorganism communities.These communities,in turn,play critical roles in host digestion,energy,and health (Lynch and Pedersen,2016;Rooks and Garrett,2016) and display complex microbial-microbial and host-microbial relationships (Wang et al.,2018).
Representing more than half of all vertebrate species,fish are an important class of animal for testing mutualistic symbiosis of microorganisms with their hosts (Talwar et al.,2018).Intestinal microbe often participates in the physiological and biological functions of their fish hosts (Rooks and Garrett,2016;Blander et al.,2017).Although such microorganisms include many categories,including bacteria,viruses,and fungi,most of our knowledge on the intestinal microbiota derives from studies on bacterial studies,with few reports on viruses and fungi.As primary decomposers of organic material,fungi including molds and yeasts are a diverse and ubiquitous group of eukaryotic organisms that contain cell walls but no chlorophyll or plastids.They inhabit both terrestrial and aquatic environments,including within the bodies of vertebrates (e.g.intestinal segments) and substantially contribute to global biogeochemical cycling (Gadd,2007;Nazir et al.,2017).Just like intestinal bacteria,fungi are an important part of the fish microbial community and play important roles in host health and disease.In aquaculture,various yeasts have been identified and studied as a part of the normal microbiota in fish intestines (Gatesoupe,2007;Nayak,2010;Sarlin and Philip,2011;Øverland et al.,2013;Caruff o et al.,2015).Thus,understanding the composition and diversity of intestinal fungal communities is essential to promote fish health and productivity.However,previous studies on intestinal fungi in fish have primarily focused on culture-based methods,with the composition and diversity of fungal communities rarely reported.
Tilapia (Oreochromismossambicus) is a tropical,salt-tolerant,omnivorous fish native to Africa,and it shows a good resistance to hypoxia and strong adaptability.Bighead carp (Aristichthysnobilis) is a unique filter-feeding freshwater fish native to China.Both species are considered economically important and are commonly found in rivers,lakes,and reservoirs of China.Although,intestinal bacteria in tilapia and bighead carp have been researched intensively (Borsodi et al.,2017;Wang et al.,2017;Hassaan et al.,2018;Li et al.,2018a,b,c;Zheng et al.,2018;Yu et al.,2019),studies on the composition,diversity,and role of intestinal fungi in these two fish remain limited.Such studies would improve our understanding of the composition and structure of microbes in fish intestines,as well as the ecological function of fish in different water bodies and their healthy ecological farming.
In this study,the intestinal fungi of tilapia and bighead carp from the same habitat (Hongchaojiang Reservoir,Guangxi,China) were investigated by Internal Transcribed Spacer (ITS) high-throughput sequencing.We hypothesized that the intestinal fungi would exhibit significant differences in composition and structure between these two fish,i.e.,species specificity,and among different intestinal segments related to their respective intestinal physiological functions.Thus,we aim to examine and compare the composition and diversity of fungal communities in different intestinal segments of tilapia and bighead carp;and clarify the functions and roles of fungi in the fish intestine.This research shall provide a theoretical basis for further studies on intestinal microbiota.
2 MATERIAL AND METHOD
2.1 Animal and sample preparation
Using net cages (mesh size 0.7 cm),three individual fish of each species (average weight of~0.5 kg) were captured randomly.After the anesthetization with tricaine methane sulfonate (MS-222) (60 mg/L,immersion for 2–3 min),the intestines of each fish were removed aseptically from the abdominal cavity using a sterile scalpel.Microbial samples were collected from the fore-,mid-and hind-intestinal segments by gently abrading the intestinal wall with a sterile spatula,with the obtained contents placed into separate 2.0-mL cryo tubes.Based on the six individual fish and three intestinal samples,we obtained 18 microbial samples in total.Upon collection,all samples were rapidly frozen in liquid nitrogen followed by storage in a -80-°C refrigerator awaiting DNA extraction.All experiments were conducted under the Guide for the Care and Use of Laboratory Animals in China.The procedures were approved by the Animal Ethics Committee at the South China Agricultural University,China (approval ID:SYXK-2019-0136).
2.2 DNA extraction,polymerase chain reaction(PCR),and sequencing
In accordance with manufacturer provided procedures,DNA was first extracted from the microbial samples using an EZNA stool DNA Kit (Omega Biotek,USA).From the resulting ITS rDNA,the ITS2 region was PCR-amplified with specific ITS primers(KYO2F:GATGAAGAACGYAGYRAA;ITS4R:TCCTCCGCTTATTGATATGC).The PCR procedures(i.e.,95 °C for 2 min;27 cycles at 98 °C for 10 s,62 °C for 30 s,and 68 °C for 30 s;final extension at 68 °C for 10 min) were conducted in 5 μL of 10× KOD buffer,1 μL of KOD polymerase,5 μL of 2.5 mmol/L dNTPs,100 ng of template DNA,1.5 μL of each primer(5 μmol/L),and ddH2O to a volume of 50 μL (Toyobo,Japan).The purified PCR amplicons were combined in equimolar amounts and sent to Gene Denovo Biological Technology Co.,Ltd.(Guangzhou,China) for pairedend sequencing (2×250,Illumina HiSeq 2500,USA).
2.3 Sequence assembly and taxonomic classification
Under specific criteria i.e.,10-bp minimum overlap and 2% mismatch error rate,the raw sequences were cleaned and assembled into raw tags using FLASH(v1.2.11) (Magoč and Salzberg,2011),followed by QIIME (v1.9.1)-based filtering to acquire clean tags(Caporaso et al.,2010;Bokulich et al.,2013).These clean tags were searched using the reference database,with chimeric tags identified and then removed using QIIME (v1.9.1).The resulting effective tags were then binned into operational taxonomic unit (OTU)clusters (≥97% similarity) with UPARSE (Edgar,2013).The absolute and relative abundances of the obtained OTUs were determined for the following analyses.All raw data were submitted to the National Center for Biotechnology Information (NCBI)Sequence Read Archive (SRA) under accession number SRP239684.
2.4 ITS fungal community analysis and functional prediction
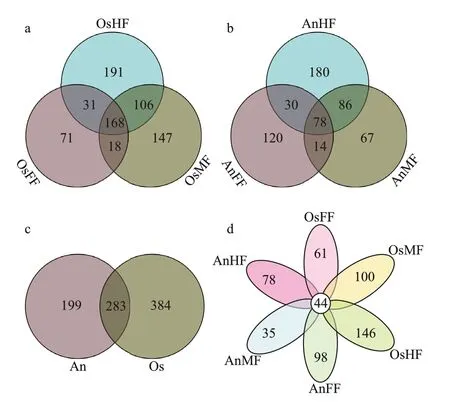
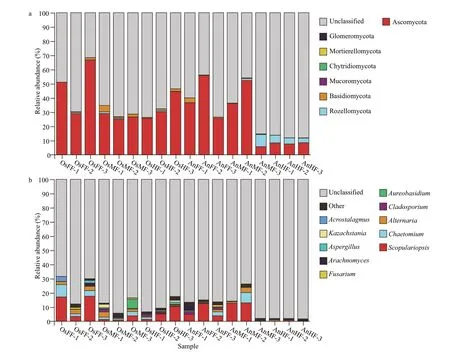
For each microbial sample,Chao1,Sobs,Shannon,and Simpson indices were calculated to determine the overall (mean) alpha diversity for each sample (with 95% confidence interval),which was then evaluated against all other microbial samples collected from the same site.Beta diversity of samples was also determined and used for principal component analysis(PCA).Nonmetric multidimensional scaling (NMDS)was evaluated and plotted using R software.The Bray-Curtis dissimilarity index was applied for analysis of similarities (ANOSIM) to establish statistical differences in the microbial community and community overlap (i.e.,R>0.75,well-separated community;0.50 From the above OTU abundances,FUNGuildbased functional annotation of the identified fungi was performed (http://funguild.org).In brief,the FUNGuild database was used for functional predictions of the OTUs of interest.From the database,annotations are classified as ‘highly probable’ or‘probable’ and include trophic mode,guild,and growth morphology.The trophic fungal guilds include pathotrophic,symbiotrophic,and saprotrophic (i.e.,procuring nutrients by damaging live host cells,exchanging resources with live host cells,or destroying dead host cells,respectively) (Nguyen et al.,2016).Species can be classified as single or multimode.Guild annotations include animal pathogens,fungi (e.g.,arbuscular mycorrhizal,ectomycorrhizal,ericoid mycorrhizal,lichenicolous,lichenized,and foliar endophytic fungi),mycoparasites,and plant pathogens (e.g.,root endophytes,saprotrophs,and wood saprotrophs).Species can be single or multiguild.Growth morphology annotations include yeast,facultative yeast,and thallus. A total of 1 763 478 high-quality sequences (tags)were obtained from the 18 samples.The number of sequences per sample ranged from 81 827 to 121 770,which clustered into 1 089 OTUs.As shown in the Venn diagrams,we obtained a total of 667 OTUs for tilapia and 482 OTUs for bighead carp (Fig.1c &Supplementary Table S1),of which 283 are overlapped between the two species (Fig.1c) and 44 overlapped in the fore-,mid-,and hind-intestine of tilapia and bighead carp (Fig.1d).In detail,we identified 288,439,and 496 OTUs for the fore-,mid-,and hindintestine of tilapia (OsFF,OsMF,and OsHF),respectively,and 242,245,and 374 OTUs for the fore-,mid-,and hind-intestine of bighead carp (AnFF,AnMF and AnHF),respectively (Fig.1a &b).A summary of the OTUs and tags of all samples are shown in Supplementary Table S1.The Venn diagrams(Fig.1c &d) indicated that many intestinal fungi were shared not only within the different intestinal segments of one fish but also among the different fish.Among the 44 overlapping OTUs (Fig.1d),14 were annotated,including 13 belonging to Ascomycota and one belonging to Rozellomycota. Fig.1 Venn diagrams showing OTUs of intestinal fungi in tilapia and bighead carp Taxonomy stack distributions were determined to analyze the composition of each sample at the phylum and genus levels.Among the annotated OTUs,the top 10 with abundances ≥2% in the sample were selected,with others classified into the“other”category and unannotated OTUs classified into the“unclassified”category (Fig.2).At the phylum level (Fig.2a),the taxonomic compositions of OsFF,OsMF,and OsHF showed relatively high average abundances of Ascomycota (48.93%±19.20%,26.88%±2.02%,and 33.40%±9.91%,respectively),followed by Rozellomycota (0.26%±0.38%,0.60%±0.59%,and 0.38%±0.33%,respectively) and Basidiomycota(0.82%±0.77%,2.46%±1.79%,and 1.21%±0.61%,respectively),whereas the composition in AnFF,AnMF,and AnHF showed a very high dominance of Ascomycota (39.30%±15.32%,31.39%±23.70%,and 8.15%±0.47%,respectively),followed by Rozellomycota (0.06%±0.06%,3.26%±4.60%,and 4.26%±1.04%,respectively) and Basidiomycota(1.46%±1.70%,0.27%±0.27 and,0.06%±0.05%,respectively).At genus level (Fig.2b),the taxonomic compositions of OsFF,OsMF,and OsHF and AnFF,AnMF,and AnHF showed relatively high average abundances ofScopulariopsis(12.68%±8.06%,2.03%±1.59%,5.62%±4.53%,7.07%±4.71%,8.57%±7.39%,and 0.39%±0.40%,respectively),Chaetomium(4.90%±3.51%,1.84%±1.07%,0.65%±0.71%,1.08%±1.48%,2.42%±4.19%,and 0.11%±0.17%,respectively),andAlternaria(2.76%±0.54%,1.97%±1.57%,0.49%±0.25%,0.60%±0.82%,1.61%±1.80%,and 0.37%±0.46%,respectively). Fig.2 Relative fungal abundances and species composition at phylum (a) and genus (b) levels based on taxonomy stack distributions It is worth noting that,Alternaria(Dothideomycetes),Scopulariopsis(Sordariomycetes),andChaetomium(Sordariomycetes) all belong to Ascomycota.Among the 1 089 fungal OTU sequences,404 were annotated,including 310 in Ascomycota,68 in Basidiomycota,three in Chytridiomycota,one in Glomeromycota,six in Mortierellomycota,three in Mucoromycota,and 13 in Rozellomycota.The dominant fungi in the intestines of tilapia and bighead carp were Ascomycota at the phylum level,which dominated in most segments and accounted for approximately one third of relative fungal abundances,andScopulariopsis(Ascomycota)at the genus level. For alpha diversity (Chao1,Sobs,Shannon and Simpson indices),no significant differences were observed between the two species (Fig.3a) or within tilapia (OsFF,OsMF,and OsHF) (Fig.3b).Within bighead carp (AnFF,AnMF,and AnHF),however,fungi in the hind-intestine differed markedly from those in the fore-and mid-intestine (Fig.3b). Fig.3 Differences in alpha diversity of intestinal fungi between (a) and within (b) tilapia and bighead carp For beta diversity,the NMDS (Fig.4) results show that tilapia and bighead carp are closely clustered but exhibit considerable differences in intestinal fungi.Pairwise ANOSIM (Table 1) indicates no significant differences among the fore-,mid-,and hind-intestinal segments within tilapia and bighead carp (P>0.05).The fungal communities of tilapia and bighead carp were significantly different but strongly overlapping(ANOSIM-R=0.415,P=0.001),which is consistent to the Venn diagram results,and shows that tilapia and bighead carp shared many intestinal fungi. Fig.4 Beta diversity of intestinal fungi within and between tilapia and bighead carp Based on the FUNGuild database,only 154 of the 1 089 OTU sequences (404 annotated OTUs) were considered reliable (probable or highly probable) and were classified into three trophic modes and nine guilds (Supplementary Table S2).The three trophic modes consisted of 108 OTUs of saprotrophic fungi,41 OTUs of pathotrophic fungi,and five OTUs of symbiotrophic fungi.Among the 108 OTUs ofsaprotrophic fungi,80 belonged to 31 known genera of Ascomycota,25 belonged to 22 known genera of Basidiomycota,and three belonged to two known genera of Mucoromycota.In addition,83 pathotrophic fungi were undefined pathotrophic,14 were wood saprotrophic,nine were dung saprotrophic,and two were soil saprotrophic.Among the 41 OTUs of pathotrophic fungi,35 belonged to 24 known genera of Ascomycota and six belonged to five known genera of Basidiomycota.In addition,23 OTUs of pathotrophic fungi were from plant pathogens and three were from animal pathogens,which may have entered the intestines via the food consumed by the fish.Among the five OTUs of symbiotrophic fungi,two belonged to two known genera of Ascomycota,two belonged to two known genera of Basidiomycota,and one belonged to one unclassified genus of Glomeromycota. Table 1 Pairwise ANOSIM of different groups based on Bray-Curtis dissimilarity index For the fore-,mid-and hind-intestinal segments of tilapia and bighead carp,the majority of taxa (relative abundance) were classified as dung saprotrophicundefined saprotrophic-wood saprotrophic(17.57%±10.85%,3.87%±2.64%,6.27%±4.05%,8.15%±4.19%,10.98%±10.21%,and 0.50%±0.56%,respectively),undefined saprotrophic (7.08%±5.83%,5.99%±5.42%,5.27%±4.11%,8.08%±0.67%,6.84%±4.58%,1.20%±0.53%,and respectively),and animal pathogen-endophyte-plant pathogen-wood saprotrophic guilds (2.76%±0.54%,1.97%±1.57%,0.49%±0.25%,0.60%±0.82%,1.61%±1.80%,and 0.37%±0.46%,respectively) (Supplementary Fig.S1).For distribution of the relative abundance mentioned above,a heatmap was constructed based on guild assignments for the top 15 fungal species determined using FUNGuild.From top to bottom,the abundance of guild assignments decreased gradually.As shown in Fig.5a,there were relatively smaller differences among OsFF,OsMF,and OsHF,but relatively greater differences among AnFF,AnMF,and AnHF.This was especially obvious for the top four guild assignments (dung saprotrophic-undefined saprotrophic-wood saprotrophic,undefined saprotrophic,animal pathogen-endophyte-plant pathogen-wood saprotrophic guilds).The PCA results for the predicted function of fungi in tilapia and bighead carp showed that fungi in the fore-,mid-and hind-intestinal segments functioned differently(Fig.5b &c).For specific annotated OTUs,the three most abundant (i.e.,Otu000002,Ascomycota;Sordariomycetes;Microascales;Microascaceae;Scopulariopsis;Scopulariopsis acremonium;Otu000018,Ascomycota;Dothideomycetes;Pleosporales;Pleosporaceae;Alternaria;Alternariapalandui;Otu000034,Ascomycota;Dothideomycetes;Dothideales;Dothioraceae;Aureobasidium;Aureobasidiumpullulans) were classified as dung saprotrophic (Otu000002) and pathotrophicsaprotrophic-symbiotrophic (Otu000018 and Otu000034),and all showed lower abundance in the hind intestine of bighead carp,in accordance with the taxonomy stack distributions.Notably,compared to the above three OTUs,many of the other annotated OTUs were detected in only one of the three segments in tilapia and bighead carp,suggesting that these corresponding fungi might exhibit specific colonization. Fig.5 The heatmap and principal component analysis (PCA) of intestinal fungi in tilapia and bighead carp In overall,our results show that,for reliable OTUs,more than two thirds were saprotrophic fungi,less than one third were pathotrophic fungi,and only 3.2%were symbiotrophic fungi;and the distribution of fungi in the intestine was uneven,especially in the hind-intestine of bighead carp,where saprotrophic and pathotrophic fungi decreased sharply,suggesting that they may be involved in digestion and absorption. In this study,Ascomycota fungi (phylum level)were dominant in the intestines of tilapia(48.93%±19.20%) and bighead carp(39.30%±15.32%).Previous research on the woodeating fishPanaquenigrolineatusalso showed Ascomycota (i.e.,Sordariomycetes and Dothideomycetes) to be dominant in the gut (Marden et al.,2017).Ascomycota fungi are heterotrophs that obtain nutrients from dead or living organisms,often forming symbiotic relationships with algae,plants,and even arthropods (Yang et al.,2012).The Ascomycota accounts for more than 75% of all fungi and contain a wide variety of organisms,including yeasts,penicillin (Penicilliumchrysogenum),morels,truffl es,and most animal and plant pathogens,and thus play a large role in the recycling of dead plant material (Schoch et al.,2009;Aranda,2016;Nguyen et al.,2017;Teixeira et al.,2017;Wijayawardene et al.,2018).For the potential functions of fungi based on the FUNGuild database,most reliable OTUs(>2/3) were saprotrophic fungi.Saprotrophic fungi primarily regulate carbon,nitrogen,and phosphorus in nutrient cycling,and represent key agents of nutrient redistribution in ecosystems (Crowther et al.,2012;Talbot et al.,2013).Notably,althoughS.acremoniumis reported to cause invasive infection in humans (Ellison et al.,1998),it has been identified as a dominant species in fore-,mid-and hind-intestinal segments in tilapia and bighead carp,suggesting that it may play a role in nutrition and absorption in the intestine due to its saprophytic ability.Thus,given its dominance,further studies on the effects ofS.acremoniumon intestinal function are required. Tilapia and bighead carp showed significant differences in the composition and structure of the intestinal fungal microbiota,which may be due to the different feeding habits or trophic level of these two fish.From a feeding habit/diet perspective,tilapia fish are omnivorous,inhabits the lower and middle water layers,and dominated by plant-based bait under natural conditions such as the lakes and reservoirs.In contrast,filter-feeding bighead carp reside in the upper and middle water layers,and mainly eat zooplankton and protozoa such as rotifers,branch horns,copepods.Therefore,it is not surprising that the tilapia fish had more OTU and higher fungal richness,which supports the view that intestinal microbial diversity usually increases sequentially corresponding to carnivores,herbivores and omnivores (Kashinskaya et al.,2018).From the perspective of function,different intestinal segments in fish exhibit different functions such as digestion and absorption (Denstadli et al.,2004;Le et al.,2019;Lin et al.,2019;Zhou et al.,2019).In this study,whether for alpha diversity or distribution of abundant specific OTUs,no significant differences in fungi was found among the three intestinal segments,but obvious differences were found between the hindintestine and the fore-and mid-intestines in bighead carp.For example,compared to that in the fore-and mid-intestines,saprotroph abundance decreased drastically in the hind-intestine of bighead carp.These results suggest that fungal community was not only related to host species specificity but also to the respective physiological functions of different intestinal segments. It is worth mentioning that 41 OTUs were annotated as probable or highly probable pathotrophic fungi in this study,implying that the intestinal mucosa may be a potential site of pathogenic invasion and colonization by fungi.Although the proportion of fungi/mycobiome in the microbiome is relatively small,i.e.,less than 0.1% in the human microbiome (Qin et al.,2010),as microbial saprotrophs,pathogens and mutualists,the fungal community plays a vital role in the health and disease of animals,plants,and ecosystems (Cui et al.,2013;Seed,2014;Peay et al.,2016;Nash et al.,2017).Thus,our study highlights the importance of investigating the diversity of fungi and their relationship with host health. Community composition and structure of the intestinal fungi of tilapia and bighead carp were comprehensively analyzed based on ITS sequencing.Our results demonstrated significant differences in the community of intestinal fungi in tilapia and bighead carp but not among their different intestinal segments.For the potential functions of OTUs in the two fish species,saprotrophic fungi were dominant,followed by pathotrophic fungi,although distribution were uneven,especially in the hind-intestine of bighead carp.This study should improve our understanding of the composition and function of fungi as an important part of the intestinal microbiota of tilapia and bighead carp. All raw data were submitted to the NCBI Sequence Read Archive (SRA) under accession number SRP239684.The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.3 RESULT
3.1 Fungal composition in tilapia and bighead carp intestines
3.2 Diversity and potential functions of fungi in intestines of tilapia and bighead carp




4 DISCUSSION
5 CONCLUSION
6 DATA AVAILABILITY STATEMENT
 Journal of Oceanology and Limnology2021年5期
Journal of Oceanology and Limnology2021年5期
- Journal of Oceanology and Limnology的其它文章
- Screening of stable internal reference genes by quantitative real-time PCR in humpback grouper Cromileptes altivelis*
- Effect of fasting on protein metabolism in muscle tissue of Larimichthys crocea revealed by transcriptome and proteome*
- Mitochondrial phylogenomics reveal the origin and adaptive evolution of the deep-sea caridean shrimps (Decapoda:Caridea)*
- C17-fengycin B,produced by deep-sea-derived B acillus subtilis,possessing a strong antifungal activity against Fusarium solani*
- Relationship between morphospecies and microcystinproducing genotypes of Microcystis species in Chinese freshwaters*
- Size-dependent spatio-temporal dynamics of eukaryotic plankton community near nuclear power plant in Beibu Gulf,China*