
超高效液相色谱-高分辨质谱法测定肉类特征肽
2021-10-09 01:34:18王忠合李晓婷胡文梅王军
食品与发酵工业 2021年18期
王忠合,李晓婷,胡文梅,王军
(韩山师范学院 食品工程与生物科技学院,广东 潮州,521041)
近年来,随着肉类价格差异不断扩大,不法商家为了追求利益,借机用价格低廉的鸡肉、马肉、鸭肉、鼠肉等肉类品种伪制牛羊肉制品,该行为会影响饮食安全。这些与价格、原料和宗教等有关的食品掺假事件,不仅侵害了消费者的权益和食品公平交易,还严重威胁到了人们的身体健康和宗教信仰[1-3]。未标识的食品成分往往成为潜在的安全隐患,如过敏原及有毒成分会影响消费者的健康,错误的标签也可能会违背特定群体的饮食习惯。对食品的种类、优劣、真伪的鉴别,尤其是对肉类的鉴别,均依赖于可靠的检测方法,基于高分辨质谱的蛋白质组学方法检测蛋白或多肽,具有灵敏度高、稳定性强等优点,可以实现多物种的同时测定,并且能够对物种的特征蛋白或多肽进行明确鉴定,在鉴别肉类掺假等领域中具有广泛的应用范围。
采用质谱法鉴别肉种属的关键技术就是特征肽段的筛选,但部分潜在的特征肽段在色谱洗脱中出峰时间太早或太晚甚至残留在柱上,在质谱检测中部分特征肽常常以相对低的丰度存在,而且食品样品的基质复杂,高丰度的非目标冗余肽段及盐等干扰物质的存在会严重影响质谱信号,加之基质的抑制作用,通过质谱检测复杂的食品样品中特定的肽段仍然是一个巨大的挑战。采用高分辨质谱法筛选潜在的特征肽,三重四极杆质谱法定量测定同位素标记或未标记的肽为肉类掺假检测提供了强有力的支撑,但是高分辨质谱法筛选出的潜在特征肽并不是都适合作为标记肽,如采用高分辨质谱法筛选到7种潜在的特征肽,在三重四极杆质谱中的多重反应监测法(multiple reaction monitoring,MRM)采集检测中只有4种特征肽可检测到[4],采用MRM法检测时需要保证特征肽可检测出,信号丰度较好且专属性强。
高分辨率质谱(highresolution mass spectrometry,HRMS)已在多个研究领域得到广泛应用,在目标物筛查以及杂质鉴定中,HRMS的使用对定性筛查和结构确认产生了革命性影响。虽然三重四极杆质谱仪仍是定量分析的“黄金标准”,但是随着HRMS技术的发展,其选择性、动态范围、线性、灵敏度等有了很大的提升,从而提高了HRMS在定量分析中的应用。在复杂基质干扰物存在的情况下,三重四极杆质谱仪法将面临极大挑战,而有着与之显著不同特性的HRMS数据采集则具有出色的选择性和灵敏度,从而成功实现定量分析。飞行时间质谱平台功能强大的数据采集模式、灵活性和出色的采集速度为科学家们的研究提供极大帮助,特别是具有目标增益功能的飞行时间质谱的准多重反应监测模式(time-of-flight mass spectrometry-pseudo-multiple reaction monitoring,TOF-MRM)。在这种新的数据采集模式下,四极杆选择母离子,并选择性地在碰撞室中进行碎裂,TOF可选择性的增加目标碎片的占空比,提高响应和选择性。TOF-MRM模式可用于完整离子(母离子 > 母离子,未使用碰撞能)或碎片离子(母离子 > 碎片离子,使用碰撞能)的采集,HRMS平台定量分析的选择性和灵敏度可大幅提升,从而提高HRMS定量分析性能[5-8]。TOF-MRM的定量分析功能让科学家们可以使用类似MRM的工作流程进行定量分析,拓展了HRMS平台在定量分析中的应用,不仅可以进行常规的定性分析,更可直接应用于低检出限(limit of detection,LOD)的定量分析。
本实验选取4种肉类中的8条特征肽为目标物,研究超高效液相色谱串联四极杆飞行时间质谱法(ultra-high pressure liquid chromatography-quadrupole time-of-flight mass spectrometry,UPLC-QTOF MS)检测参数、色谱分离条件、预处理等参数对特征肽检测的影响,以期建立高效率、高灵敏的肉类制品中特征肽的检测方法,为建立肉类掺假的检测方法提供数据和理论支持。
1 材料与方法
1.1 材料与试剂
胰蛋白酶(牛胰腺来源,10 000活力单位/mg蛋白)、正己烷(色谱级)等,美国Sigma-Aldrich公司;尿素、碳酸氢铵、硫脲、二硫苏糖醇(dithiothreitol,DTT)、碘代乙酰胺、三(羟甲基)氨基甲烷盐酸盐、牛血清白蛋白、考马斯亮蓝G250等试剂(纯度≥99%)、固相萃取空柱(3 mL),生工生物工程(上海)有限公司;质谱级乙腈、甲酸和甲醇,美国Fisher公司;氨水(质谱级),美国Waters公司;合成肽,上海强耀生物科技有限公司;超纯水(≥18 MΩ·cm),实验室自制;牛肉、猪肉、鸡肉、鸭肉,当地超市;肉丸制品,当地市场;亮氨酸脑啡肽(质谱级)、CAX阳离子交换小柱(3 mL/200mg)、亲水/亲油平衡(hydrophile-lipophile balance,HLB)固相萃取柱(3 mL/200mg),美国沃特世有限公司;N-丙基乙二胺键合固相萃取填料、ENVI-18固相萃取填料,美国Sigma-Aldrich公司。
1.2 仪器与设备
Waters I-Class Plus超高效液相色谱仪(由二元泵、柱温箱、自动进样器、脱气机等组成)、Xevo G2-XS QTOF高分辨质谱仪[配电喷雾电离源(electrospray ionization,ESI)],美国沃特世有限公司;JY3002电子天平,美国梅特勒-托利多仪器有限公司;DZF-6050型真空干燥箱,巩义市予华仪器有限责任公司;ZX4漩涡振荡器,意大利VELP公司;JSP-200型高速多功能粉碎机,浙江省永康市金穗机械制造厂;Milli Q超纯水机,美国密理博有限公司。
1.3 实验方法
1.3.1 蛋白提取
先将原料肉搅碎,再称取0.5 g肉样于10 mL离心管中,加入4 mL提取缓冲液(0.3 mol/L KCl、0.3 mol/L KH2PO4、pH 6.5,或6 mol/L尿素、2 mol/L硫脲、50 mmol/L Tris-HCl),用旋涡混合器混匀5 min,超声处理30 min,在室温下于10 730×g离心40 min,收集上清液,转移到10 mL离心管中并涡旋1 min。随后,将提取物在室温下1 315×g离心3 min,以消除先前步骤中可能产生的泡沫。该上清液即为牛肉蛋白原样液,采用考马斯亮蓝染色法测定蛋白含量,于4 ℃下保存。
1.3.2 蛋白酶解
取0.1 mL提取液用0.4 mL缓冲液混合稀释加入到试管中,然后加入2.4 mL浓度为30 mmol/L 碳酸氢铵混合均匀,将提取物在90 ℃下加热30 min,以使溶液中的蛋白质变性,冷却至室温,随后添加50 mmol/L 二硫苏糖醇使其最终浓度为5 mmol/L。添加150 mmol/L碘代乙酰胺使蛋白质烷基化,最终浓度为15 mmol/L,暗处反应30 min。随后,添加1.5 mL浓度为1 mg/mL的胰蛋白酶溶液(用30 mmol/L 碳酸氢铵配制),立即将酶解液涡旋1 min,置于37 ℃酶解12 h。
1.3.3 肽段纯化
将酶解液过固相萃取柱进行脱盐处理,先用3 mL甲醇洗涤和活化萃取柱,然后用3 mL体积分数1%的甲酸平衡。将酶解液过柱,并用0.5 mL体积分数为1%的甲酸冲洗柱子,将洗涤液装入小柱,以定量转移样品。然后用1 mL体积分数为1%的甲酸洗涤小柱。最后,将肽用3 mL的乙腈-水混合物(体积比1∶1,含体积分数0.1%甲酸)洗脱到离心管中,分2次重复洗脱,第1次加2 mL混合洗脱液,第2次加1 mL混合洗脱液,在第1次洗脱液后,将小柱浸泡10 min以使肽完全洗脱[9]。随后,将全部洗脱液在氮气流中吹干,分析前将纯化物用0.5 mL的乙腈-水混合物(体积比3∶97,含0.1%甲酸)复溶,涡旋30 s,过0.22 μm滤膜,上机检测。
1.3.4 色谱-质谱条件
色谱条件:色谱柱Acquity UPLC BEH C18柱(2.1 mm×100 mm,1.7 μm),柱温40 ℃,进样室温度10 ℃,流速0.2 mL/min;流动相A为0.1%(体积分数)甲酸-水溶液,流动相B为0.1%(体积分数)甲酸-乙腈溶液;梯度洗脱时间分别探讨15、20、30、35 min,具体洗脱程序如表1所示。

表1 液相洗脱程序Table 1 Gradient elution program employed for the separation of peptides
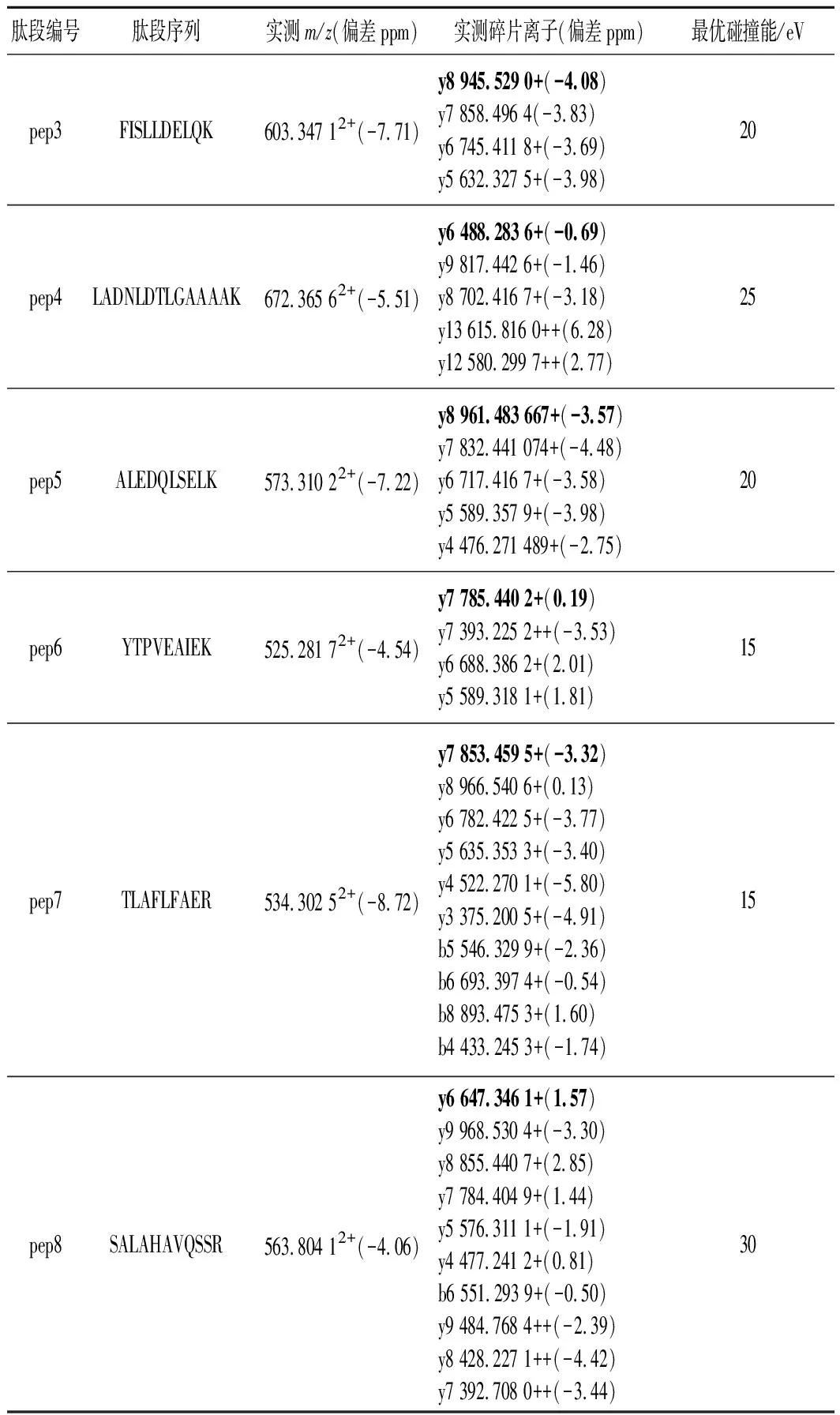
质谱条件:正离子(ESI+)灵敏度模式,毛细管电压3.0 kV,离子源温度130 ℃,脱溶剂气温度400 ℃;锥孔气流速60 L/h,脱溶剂气流速800 L/h,扫描时间0.2 s,数据格式为棒状图,一级母离子扫描范围m/z为400~1 500,二级碎片离子扫描范围m/z为50~2 000,前1 min和后6 min的流动相进废液不采集信号;MS/MS优化采集方法:根据各肽段的理论值(表2),母离子m/z分别固定设为576.32(pep1)、554.79(pep2)、603.34(pep3)、672.36(pep4)、573.31(pep5)、525.28(pep6)、534.30(pep7)、563.80(pep8),不加碰撞能;二级碎片离子碰撞能CE值分别设10、15、20、25、30、35、40 eV。TOF-MRM定量采集方法:母离子质量数分别设为384.55(pep1)、554.79(pep2)、603.34(pep3)、672.36(pep4)、573.31(pep5)、525.28(pep6)、534.30(pep7)、376.20(pep8),并选择目标增益功能;各肽段碎片定量离子和碰撞能采用优化后的值。采用质量锁定技术测定准确质量数,以200 pg/μL的亮氨酸脑啡肽溶液为锁定质量溶液,流速为5 μL/min,电压为0.53 kV,每隔30 s采集1次,在正离子模式下产生m/z556.277 1的离子[10]。

表2 TOF-MRM测定肉种属用的各特征肽段的信息Table 2 Details of marker peptides determined by TOF-MRM in meat species determination

续表2
1.3.5 基质效应(matrix effect,ME)评价
分别准确称取牛肉丸样品9份,加缓冲液(0.3 mol/L KCl、0.3 mol/L KH2PO4、pH 6.5)提取,离心后取上清液过HLB柱净化,收集流出液过0.22 μm微孔滤膜,作为样品溶液。取净化后的样品溶液1.6 mL,加入0.4 mL的混合标准肽工作溶液至终质量浓度为2.88~1 250 μg/L,注入UPLC-QTOF MS测定,分别作基质匹配和溶剂外标曲线。ME以基质匹配标准曲线的斜率与溶剂曲线斜率的比值评定[11],比值>1,表现为基质增强效应;比值<1则为基质抑制效应,用ME表示,ME计算如公式(1)所示:
(1)
1.4 数据处理
采用Waters公司的MassLynx 4.1软件,通过提取MS/MS采集方法中各通道母离子及碎片离子的质谱图,调节碰撞能量以获得各衍生产物的信号强度高、稳定性好的离子碎片,并筛选出定量离子对。TOF-MRM定量采集图选用ApexTrack的积分方法,适当调整积分参数的方式完成色谱积分,根据质谱图中碎片离子峰的强度进行定量分析[10]。实验结果以平均值±标准偏差表示,采用SPSS 17.0进行统计分析,以P<0.05为检验标准。
2 结果与分析
2.1 质谱参数的选择
分别取合成肽的母液1.0 mL,分别加入3%乙腈水(含0.1%甲酸)复溶液稀释成浓度为0.1 mg/L的肽标准溶液,将各肽标准溶液注入进样器中,色谱分离后在正离子模式下进行MS/MS扫描,得到各肽的母离子及碎片离子峰的扫描图,如图1所示。毛细管电压、锥孔电压和碰撞能量是影响质谱测定结果的重要因素,锥孔电压直接影响各组分的灵敏度[10],过高的锥孔电压会导致分子离子在离子源内发生碰撞解离,从而影响母离子的丰度,进而影响检出限和灵敏度。锥孔电压40 V且不加碰撞能量下采集的质谱图中各肽母离子带2个或3个电荷的信号强度高、稳定性好,施加碰撞能量后采集的二级质谱图中各肽段的碎片离子主要是y离子,且母离子带3个电荷的肽段所得碎片离子中伴有带2个电荷的碎片离子形成,如表3和图1-a所示。各肽段施加不同碰撞能量对碎片离子质谱图的影响如图1-b所示,随着施加碰撞能量的增大,各肽段的母离子信号强度降低、碎片离子信号强度增加,如前所述主要以y离子形式存在,分别选择576.322+(pep1)、554.792+(pep2)、603.342+(pep3)、672.362+(pep4)、573.312+(pep5)、525.282+(pep6)、534.302+(pep7)、563.802+(pep8)作为各肽段定量分析的母离子,使用TOF-MRM采集模式和目标增益采集数据,该模式下四极杆中选择母离子,仪器的目标增益功能可增加目标离子的灵敏度,从而大大增强了定量分析的灵敏度和选择性。优化后的定量分析的碎片离子和碰撞能如表3所示。
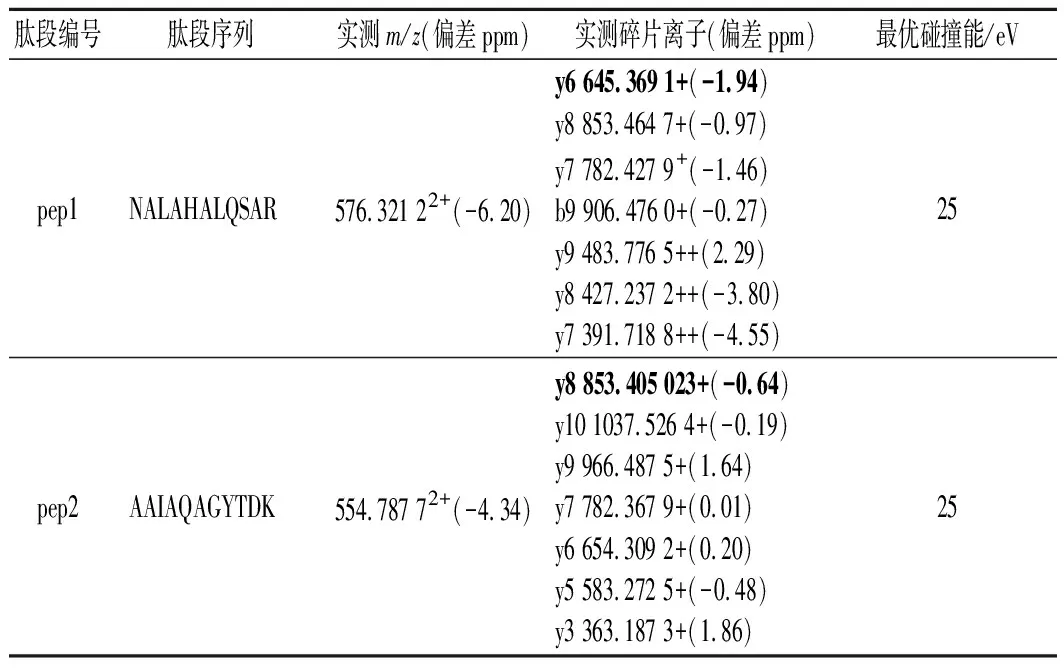
表3 八条特征肽段测定结果Table 3 Measured results of eight marker peptides
2.2 色谱条件的优化
质量浓度为0.1 mg/L的肽混合标准溶液1.0 mL,分别采用1.3.4中的3种洗脱梯度进行色谱分析,TOF-MRM质谱模式采集,检测结果如图2所示。
由图2可以看出,不同梯度洗脱分离的8种特征肽的分离效果差异较大,大部分肽的色谱图的峰形较好,各特征肽的信号强度较强,信号稳定,30 min洗脱程序的分离效果更好,但特征肽1和特征肽6的分离效果较差,分离度仅为0.99,而特征肽7和特征肽3的出峰时间较晚,分离效果非常好,因而可以在此基础上进行优化,压缩前5 min和最后10 min程序洗脱的时间,拉长中间程序洗脱的时间,优化后的洗脱程序如表4所示。

表4 优化后的8种特征肽液相色谱分离洗脱程序Table 4 Optimized elution procedure of 8 marker peptides by liquid chromatography separation
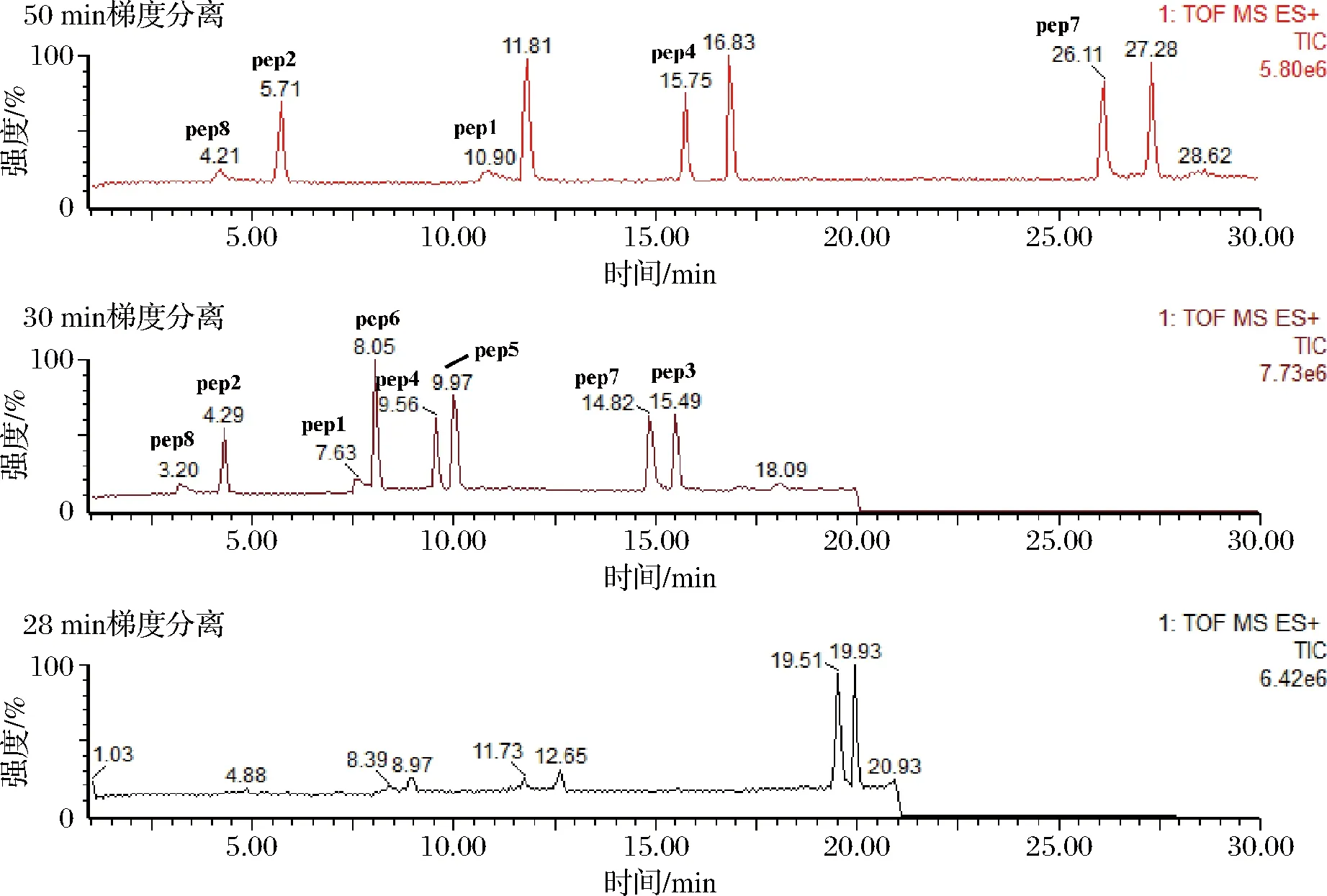
图2 液相色谱梯度洗脱程序的优化Fig.2 Optimization of gradient elution procedure in liquid chromatography
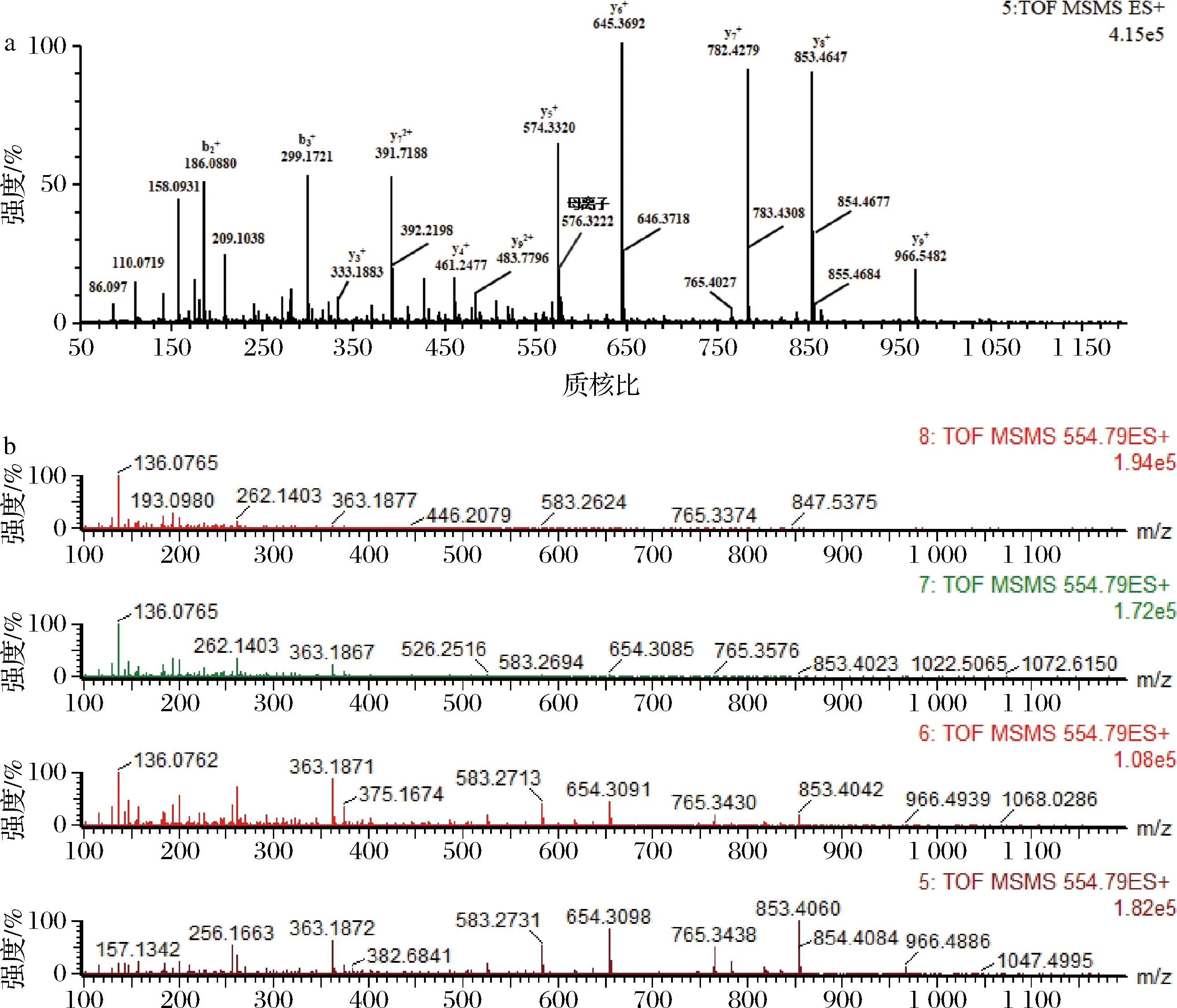

a-碎片离子形成;b-碰撞能优化图1 肽中碎片离子形成和碰撞能优化Fig.1 Fragment ion formation in marker peptides and optimization of collision energy in MS/MS acquisition modes注:图1-b中2至8通道分别施加10、15、20、25、30、35、40 eV碰撞能
2.3 质谱参数的优化
由图3可以看出,特征肽pep8和pep1的信号强度较弱,这主要是因为在MS/MS模式下设定带2价电荷的母离子,而TOF-MRM模式下pep8和pep1的母离子有带2价电荷的母离子和带3价电荷的母离子,二者共存,且带3价电荷的母离子的信号强度更大,因而特征肽pep8和pep1的母离子需要选带3价电荷的离子作为目标母离子,信号强度分别增强3.8和6.6倍,优化后的各特征肽TOF-MS/MS图谱如图4所示,分离效果好、信号稳定。
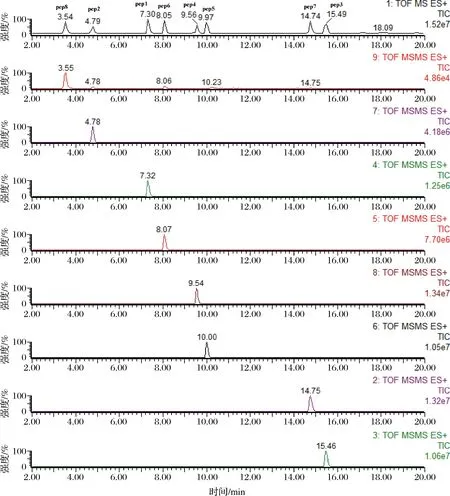
图4 优化后的肽测定图谱Fig.4 The optimized peptide chromatogram
续表3

a-特征肽测定的色谱-质谱图;b-特征肽pep1的母离子质谱图;c-特征肽pep8的母离子质谱图图3 母离子对TOF-MRM定量分析的影响Fig.3 Effect of parent ions on TOF-MRM quantitative analysis
2.4 方法学考察
移取适量的特征肽混合标准工作溶液,按照1.3的方法处理,配制系列浓度的特征肽混合标准溶液测定,各特征肽的检测结果如表5所示。由于所添加的2种特征肽为样品中的内源性物质,因此标准曲线是在扣除基质本底后绘制而成,其余特征肽可直接绘制标准曲线。LOD和定量限(limit of quantitation,LOQ)是通过在空白样品中添加目标组分的方法分别依据其特征离子色谱峰的信噪比(S/N)大于3和10而测定的[12]。8种目标特征肽的线性相关系数均大于0.98,样品中各测定的目标特征肽均在线性范围内。一般情况下,ME在85%~115%时不存在明显的基质效应。据此判断,本检测方法对特征肽1、3、4有较强的抑制效应,而对其他特征肽的基质效应不显著(表5)。
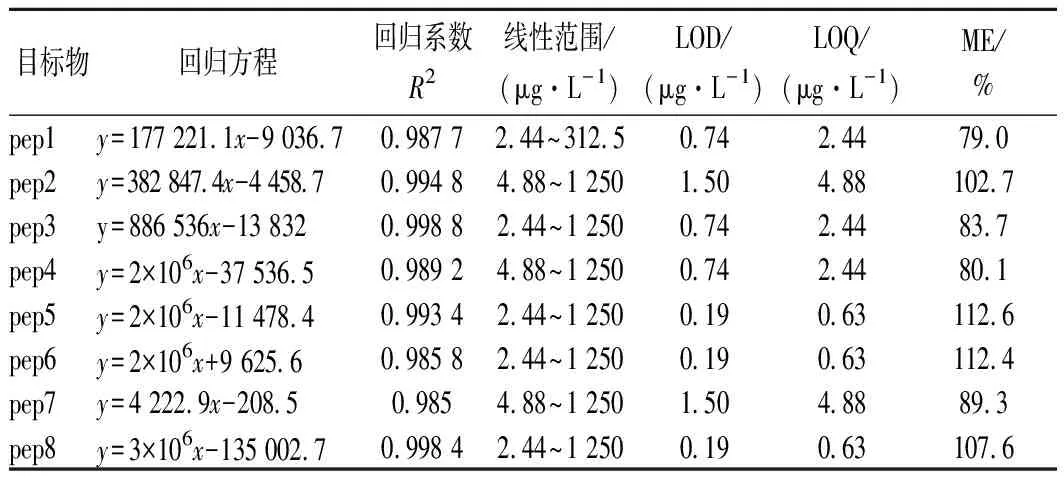
表5 特征肽测定的回归方程、检出限、定量限及METable 5 Regression equation, detection limit,quantitation linit and ME for determination of marker peptides
2.5 方法的回收率、精密度及其稳定性
移取适量的特征肽混合标准工作溶液,加入到肉样品中,配制低、中、高3个质量浓度水平(1、10、50倍定量限)的添加试验,按照1.3.1的方法处理,回收率和精密度实验测定结果如表6所示。各特征肽目标物的加标回收率为61%~102%,相对标准偏差(relative standard deviations,RSD)<10%,可用于肉及肉制品样品中特征肽含量的测定。
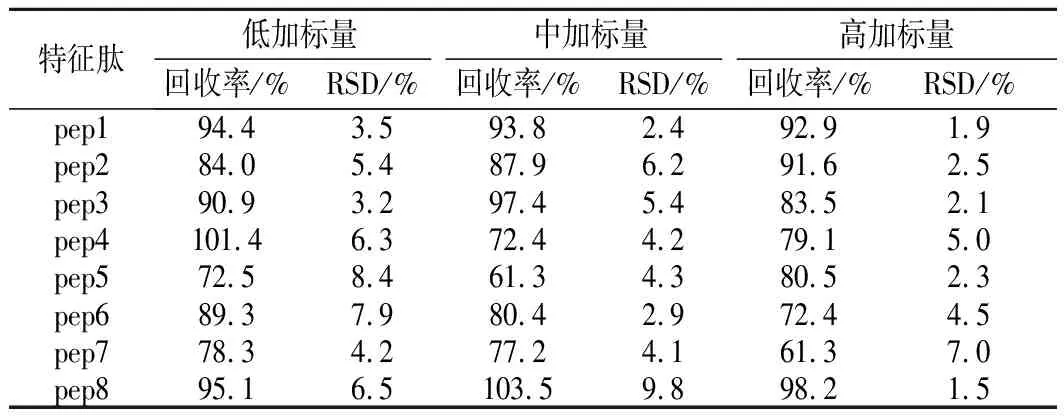
表6 样品中特征肽测定的添加回收率及RSDTable 6 Recoveries and RSD of marker peptides spiked in meat samples
2.6 实际样品的测定
为了验证所建立分析方法的适用性,对市场上随机抽取的7种价位的35份牛肉丸样品进行检测,结果如表7所示。结果显示,本次抽样筛查的牛肉丸样品中共检测出5条特征肽,其种属来源分别为牛肉2条、猪肉1条、鸡肉2条,这表明牛肉丸样品中不同程度地混有其他种类的肉,其混入量不等,与牛肉丸的价格高低无关。鸭肉中的2条特征肽没检出,可能是牛肉丸样品中未混入鸭肉,而另1条猪肉特征肽未测到,可能是与该特征肽在猪不同部位的肉中的分布有关,有待深入探讨,后续论文另有研究报道。并且采用四极杆串联轨道阱中的信息依赖性、信息非依赖性、平行反应模式等采集模式及PCR方法进行对比,测定结果与本方法吻合,进一步证明了所建方法的可靠性。
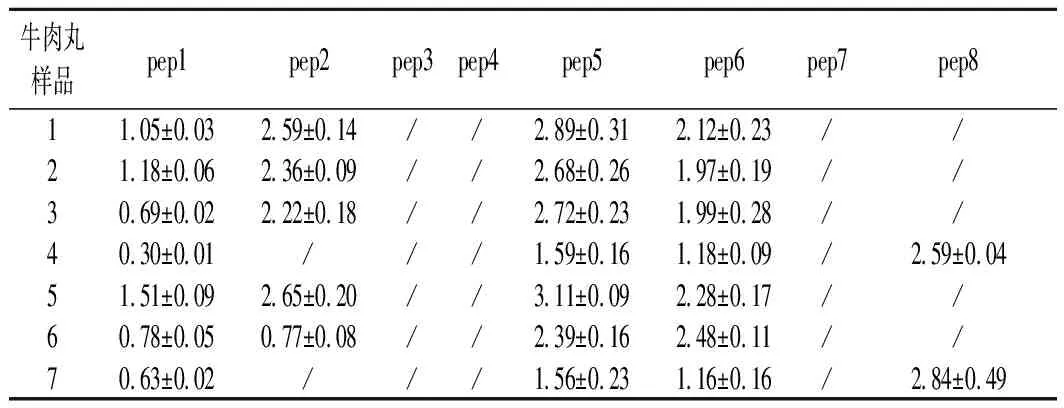
表7 牛肉丸样品中特征肽测定的结果 单位:mg/g
3 结论
8条目标特征肽的母离子带2个或3个电荷的信号强度高、稳定性好,施加碰撞能量后采集的二级质谱图中各肽段的碎片离子主要是y离子,且母离子带3个电荷的肽段所得碎片离子也有带2个电荷的碎片离子,使用TOF-MRM采集模式和目标增益采集数据可增加目标离子的检测灵敏度,从而大大增强了定量分析的灵敏度和选择性。优化后梯度对8条特征肽的洗脱分离效果非常好,特征肽8和特征肽1的母离子选带3价电荷的离子,优化后的质谱信号稳定、检出限低,相关系数在0.99左右,各特征肽目标物的加标回收率为61%~102%,RSD<10%,可用于肉及肉制品中特征肽含量的测定及肉类掺假鉴别。市场上7种价位的牛肉丸中共检出5条特征肽,其中2条来源于鸡肉、1条来源于猪肉,这表明牛肉丸样品中不同程度地混有鸡肉或猪肉,且其混入量不等、与价格高低无关,这进一步证明了所建方法的可靠性与可应用性。
猜你喜欢
食品安全导刊(2021年20期)2021-08-30 06:39:48
动漫星空(兴趣百科)(2020年4期)2020-03-26 08:08:46
黄河黄土黄种人(2019年12期)2019-02-06 04:02:15
中学生数理化·高一版(2016年7期)2016-12-07 20:47:07
试题与研究·中考化学(2016年1期)2016-09-30 18:38:21
小星星·阅读100分(高年级)(2016年4期)2016-04-28 20:30:29
当代化工研究(2016年5期)2016-03-20 16:21:35
浙江大学学报(农业与生命科学版)(2015年4期)2015-12-15 12:47:46
电源技术(2015年5期)2015-08-22 11:17:54
广东第二课堂·小学(2014年6期)2014-09-01 17:00:02
