
桑品种桂桑优12的转录组生物信息学分析*
2021-10-08 05:30:14张朝华
蚕学通讯 2021年3期
张朝华 王 霞
(广西壮族自治区蚕业技术推广站,南宁 530007)
桑树(MorusL.)作为养蚕业的饲料树种具有重要的经济价值,近10多年桑树的生态价值、药食用价值、畜禽用饲料价值等被逐渐发掘利用[1],因而桑树已受到农林业、畜牧业及医疗保健行业的关注。然而,目前桑树的基础研究还相对滞后,例如桑树对不良环境的适应能力、桑树含有的许多活性物质及药用功效等,虽然都有一定的试验证据支持,但还不能从分子水平解析其产生、形成机制。利用基因组、转录组及蛋白质组学等组学技术探讨植物的生理活动规律及生物代谢的机制[2],成为当下的研究热点。自2013年西南大学何宁佳的研究团队完成了川桑(Morusnotabilis)的基因组测序工作[3]之后,桑树功能基因组的研究也取得了重要进展[4-6]。转录组学是由Velculescu等[7]在1997年提出,了解转录组对于分析基因表达和鉴定未知基因,揭示细胞与组织的分子成分以及理解生长发育和抗性形成等是必不可少的,目前转录组学已应用于多种植物的研究[8-10]。我们拟对桑树品种桂桑优12的根部总RNA进行转录组测序,利用生物信息学软件对测序数据进行拼接组装分析,对unigene进行功能分类注释等,希望能够为桑树重要性状基因的发掘及功能分析,为桑树遗传图谱构建以及分子育种积累一定的基础数据。
1 材料与方法
1.1 桑树材料
桂桑优12为广西蚕业技术推广站育成的桑树品种,以该品种的3年生植株根部材料供试。
1.2 总RNA提取及检测
按照天根植物总RNA提取试剂盒DP432的说明书操作提取桑树根尖总RNA,经1%琼脂糖凝胶电泳检测RNA提取质量。电压180 V电压,电泳16 min,Agilent 2100检测RNA的完整性(RIN值≥6.6)。
1.3 转录组测序及测序组装
总RNA样品检测合格后,转录组测序由北京诺禾致源科技股份有限公司完成。
cDNA文库的构建:用带有Oligo(dT)的磁珠富集mRNA,随后加入fragmentation buffer将mRNA打断成短片段,以mRNA为模板;用六碱基随机引物(random hexamers)合成第1链cDNA,然后加入缓冲液、 dNTPs和DNA polymeraseⅠ和RNase H合成第2链cDNA,再用AMPure XP beads纯化双链cDNA;纯化的双链cDNA先进行末端修复、加A尾并连接测序接头,再用AMPure XP beads进行片段大小选择;最后进行PCR扩增,并用AMPure XP beads纯化PCR产物,得到最终的文库。
文库构建完成后,先使用Qubit2.0进行初步定量,稀释文库至1.5 ng/μL,随后使用Agilent 2100对文库的insert size进行检测,insert size符合预期后,使用Q-PCR方法对文库的有效浓度进行准确定量(文库有效浓度>2 nmol/L)。文库检验合格后,把不同文库按照有效浓度及目标下机数据量的需求pooling后进行Illumina HiSeq测序。
1.4 测序数据信息分析
将测序获得的转录组原始数据进行转录本拼接,拼接后用Corset程序进行聚类,以聚类后的序列进入各大功能数据库比对,进行功能注释,包括GO注释、KEGG注释和COG注释。
2 结果与分析
2.1 转录组测序及组装结果分析
对桂桑优12根尖组织的转录组测序后共获得50 844 314条原始测序数据(Raw data),原始测序数据经过滤后得到Clean data 50 540 436条。对Clean reads进行从头组装和序列去冗余后共获得了102 254个转录本,转录本的总长度、平均长度和N50值分别为64 665 080 bp,771 bp和1 473 bp。转录本的长度分布情况见图1。
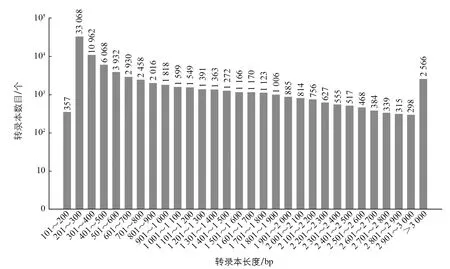
图1 桂桑优12根尖组织转录本的长度分布
通过将转录本比对到NR,NT,SwissProt,COG,GO,KEGG,PFAM,InterPro等8大功能数据库, 对转录组测定的序列进行功能注释, 其中有86 332个转录本获得了注释结果。同时,在本项目中总共预测出68 218个编码序列(Coding DNA sequence,CDS),其中通过注释结果检测出57 348个CDS,使用ESTScan预测出10 870个CDS。此外,还在27 068个转录本中检测到41 882个简单重复序列(Simple sequence repeat,SSR)。
2.2 转录本功能注释
2.2.1 KEGG功能注释
KEGG是一个生物信息学的系统数据库,其将基因组和系统功能信息等整合在一起,通过将基因在基因组或转录组的含量映射到KEGG数据库的代谢通路的过程,将基因组与有机体的系统行为连接起来[11]。本次对桂桑优12根尖组织的转录组分析中:有2 143个unigene被归到运输和分解代谢途径,599个unigene被归到膜输送途径,1 020个unigene被归到信号传导途径,3 354个unigene被归到折叠分类降解途径等(图2)。
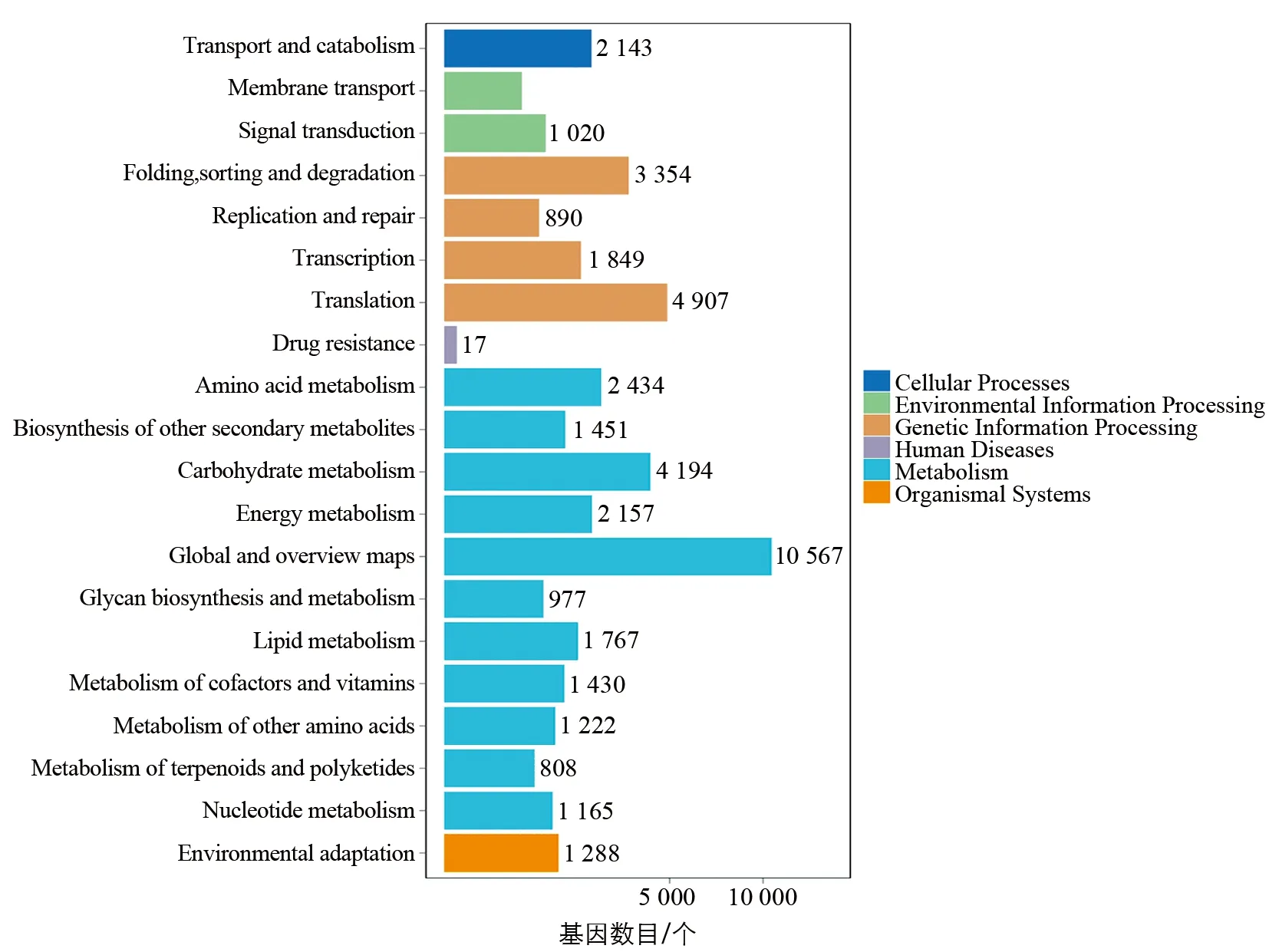
图2 桂桑优12根尖组织转录组序列的KEGG途径分布
2.2.2 COG功能注释
蛋白质直系同源簇(COGs)是通过对某些真核及原核生物等完整基因组编码蛋白质,根据生物系统的进化分类关系而构建的,可应用于预测单个蛋白质或整个新基因组中蛋白质的功能[12]。基于COG数据库可以对蛋白质进行系统进化分类的功能,将获得的桂桑优12根尖组织的转录组序列比对到COG数据库,可将这些序列分为25类(图3)。其中,有5 087个序列归为蛋白质功能预测类,有4 104个序列被归为翻译、核糖体结构及生物发生一类,有2 690个序列被归为翻译后修饰、蛋白质折叠及分子伴侣类。
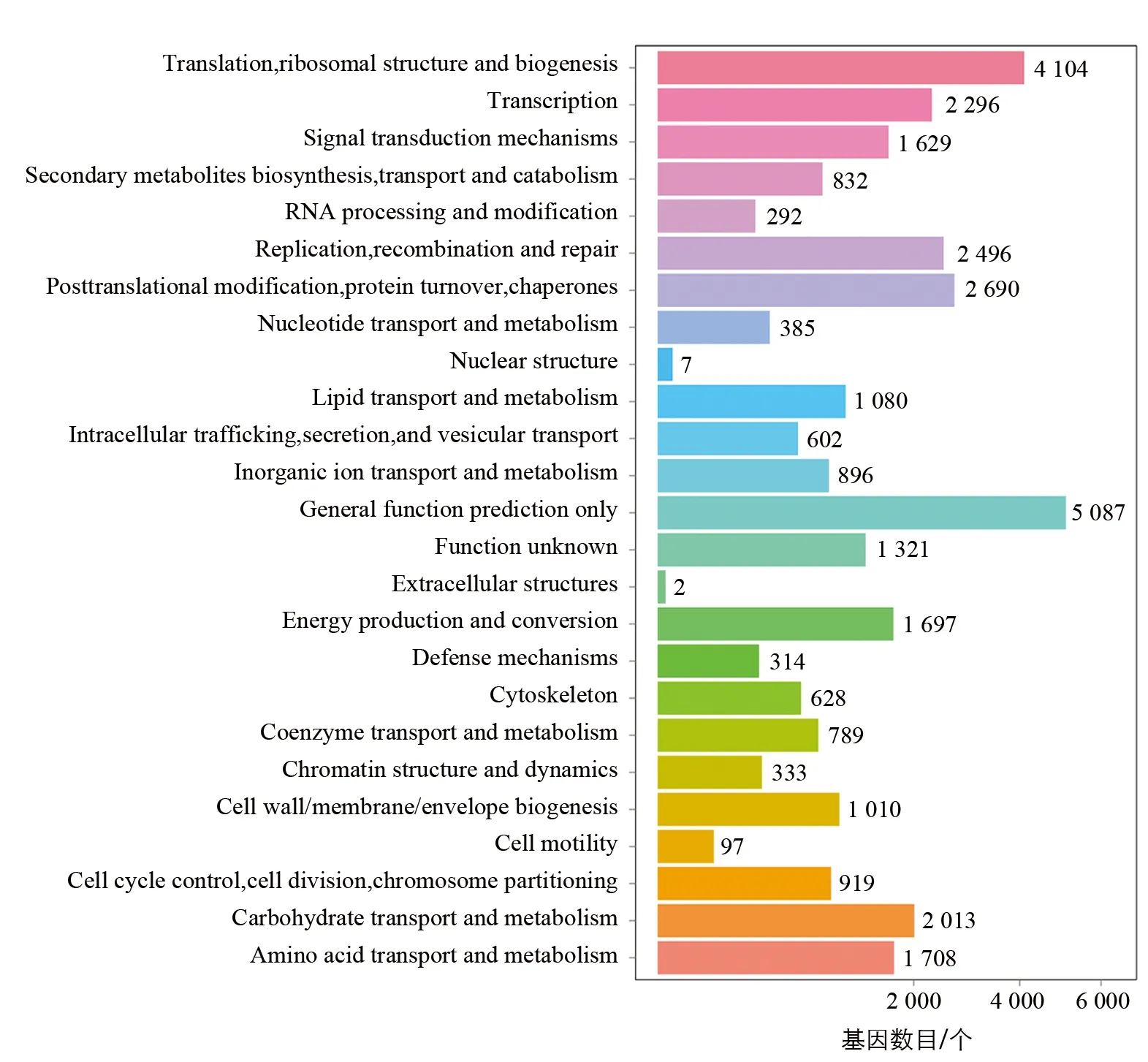
图3 桂桑优12根尖组织转录组序列的COG功能注释分布
2.2.3 GO功能注释
GO(Gene Ontology, http://www.geneontology.org)数据库将所有与基因有关的研究结果进行分类汇总,形成标准化的基因和基因产物的生物学术语,该数据库对基因和蛋白质功能进行统一的界定和描述。GO数据库从3个方面,即组建细胞成分功能(CC)、参与生物过程功能(BP)和分子生物功能(MF)等,对基因及其产物进行分类注释。桂桑优12根尖组织的转录组序列被归为55大类(图4),其中参与代谢过程类等7类的转录序列均超过10 000个,参与膜结构和生物调节类等16个分类的序列均超过1 000个,其它分类中参与的序列相对较少,最少的如翻译调节活性类仅有1个序列。
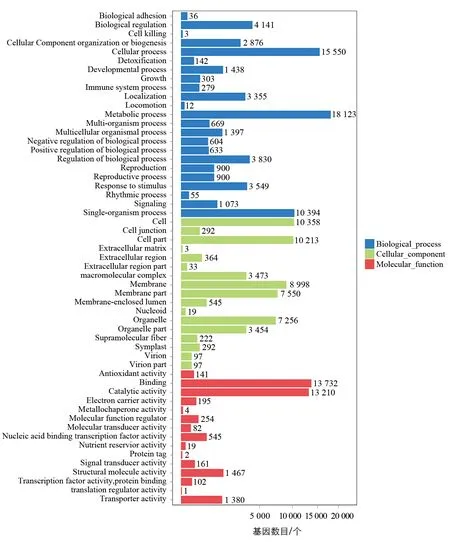
图4 桂桑优12根尖组织转录组序列的GO功能注释分布
3 结 论
本研究以广西蚕区大面积种植桑品种桂桑优12的根尖组织总RNA进行转录组测序,并将序列在KEGG,GO及COG等数据库中进行分析归类。测序共获得50 844 314条原始测序数据(Raw data),经过滤原始测序数据后获得得到99.4%的Clean data,共计50 540 436条,得到7.38 G的数据,GC含量为46.58%。对Clean Reads进行拼接组装及去冗余后得到102 254个转录本。利用COG,GO,KEGG等功能数据库对序列进行功能注释, 其中有86 332个转录本获得了注释结果,总共预测出的68 218个CDS中,有57 348个CDS获得注释。86 332个unigenes在KEGG中获得注释的有40 254个,在COG中获得注释的有26 832个,在GO中获得注释的有27 766个。本次测序获得的桂桑优12转录组数据及功能注释结果,为今后探索了解桑树的多种生理生化过程及代谢机制,挖掘桑树的特殊性状及开展桑树遗传育种等方面的研究,积累了具有一定参考意义的基础数据。
猜你喜欢
音乐教育与创作(2022年1期)2022-04-26 02:21:20
今日农业(2021年11期)2021-08-13 08:53:24
猪业科学(2021年3期)2021-05-21 02:05:36
今日农业(2020年16期)2020-12-14 15:04:59
幽默大师(2020年10期)2020-11-10 09:07:22
中华诗词(2019年1期)2019-11-14 23:33:56
猪业科学(2018年4期)2018-05-19 02:04:31
小学生作文(中高年级适用)(2018年3期)2018-04-18 01:24:40
学生天地(2016年16期)2016-05-17 05:46:06
遗传(2014年3期)2014-02-28 20:58:49
