
2 株伪狂犬病毒变异株全基因组测序及主要保护性抗原氨基酸变异分析
2021-10-05 07:55:38郭振华邢广旭翁茂洋金前跃乔松林张改平
河南农业科学 2021年8期
郭振华,邢广旭,翁茂洋,金前跃,乔松林,张改平,2,3
(1.河南省农业科学院 动物免疫学重点实验室,河南 郑州 450002;2.河南农业大学 动物医学院,河南 郑州 450002;3.江苏高校动物重要疫病与人兽共患病防控协同创新中心,江苏 扬州 225009)
伪狂犬病(Pseudorabies,PR)也称奥耶斯基氏病(Aujeszky’s disease,AD),最早由匈牙利科学家奥耶斯基(Aujeszky)于1902年报道。其致病病原是伪狂犬病毒(Pseudorabies virus,PRV),也称为猪疱疹病毒1 型(Suid herpesvirus 1,SuHV-1)和奥耶斯基病毒(AD virus,ADV)[1]。PRV 可以感染多种动物,如狗、猫、羊、牛、狐狸、貂和鼠等,致死率几乎为100%[2]。猪是PRV 唯一的自然宿主,PRV 感染后可以在猪体内建立起持续终身的潜伏感染。母猪感染后主要表现为流产、返情和死胎等繁殖障碍症状;仔猪主要表现为流涎、口吐白沫、颤抖、角弓反张等神经症状,死亡率很高[3];保育猪通常以神经症状为主,同时伴有一定的呼吸道症状,但死亡率大大降低;育肥猪则以呼吸道症状为主,偶见轻微的神经症状[4]。
从20 世纪90 年代到2010 年,我国主要通过接种PRV 弱毒疫苗(如Bartha-K61 株和HB-98 株)防控该病,并取得了良好的效果。2011 年开始,许多接种疫苗的猪场相继暴发了PR[5-6]。AN等[1]证实,该次疫情是由新出现的PRV 变异株引起的,且现有疫苗(Bartha-K61 株)针对变异株感染不能提供完全保护。随后PRV 变异株在我国多个省份迅速传播,GU等[7]于2013—2016年对山东省采集的5 033份血清样品进行检测,结果显示,PRV gE 抗体阳性率为57.8%;解伟涛等[8]于2014—2016 年针对河南地区临床样本的监测数据显示,PRV 野毒感染的猪场阳性率达到了90%左右,血清样品的gE 抗体阳性率为46.2%。PRV 变异株的再度流行给我国养猪业的发展带来了新的挑战。
gB、gC和gD 蛋白是PRV 主要的保护性抗原,与免疫保护紧密相关[9-12],研究其变异特征对监测我国PRV 毒株的遗传变异情况具有重要意义。鉴于此,对本实验室分离的PRV 毒株HeNLH/2017 和HeNZM/2017 进行全基因组序列的测定,并利用生物信息学方法分析其主要保护性抗原的遗传变异情况,以丰富我国PRV 流行毒株的基因组数据,旨在为PR的流行与防控提供参考。
1 材料和方法
1.1 毒株及细胞
HeNLH/2017 和HeNZM/2017 PRV 毒株由本 实验室从河南漯河地区和中牟地区临床病料中分离获得。病毒的扩繁在PK-15细胞上进行。
1.2 病毒培养与核酸提取
将HeNLH/2017 和HeNZM/2017 两个PRV 毒株分别接种于PK-15 细胞,待病变率达到80%时,收集培养上清并保存;贴壁细胞用PBS洗1遍,然后按照基因组提取试剂盒操作说明提取细胞和病毒的总DNA。用NanoDrop One(ThermoFisher Scientific公司)仪器测定提取核酸的浓度。
1.3 病毒基因组测序
将提取的总DNA 送至上海派森诺生物科技股份有限公司进行病毒基因组测序,采用Illumina NovaSeq 二代测序方法。序列的拼接和标注参照HN1201毒株(GenBank No.KP722022)进行。
1.4 基因组同源性分析
基因组同源性分析采用平均核苷酸相似性(Average nucleotide identity,ANI)(https://www.ezbiocloud.net/tools/orthoani)进行。
1.5 进化树及主要抗原氨基酸变异分析
从GenBank 数据库获得参考毒株的序列(表1)。全基因组 序列的比对采用MAFFT(Multiple alignment using fast fourier transform)程序进行;系统发育进化树的构建采用MEGA 6.0的邻位法(Neighbor-joining tree with 1 000 bootstrap)进行。利用DNAStar 软件中的MegAlign(Clustal W method)程序进行氨基酸序列的多重比对分析。
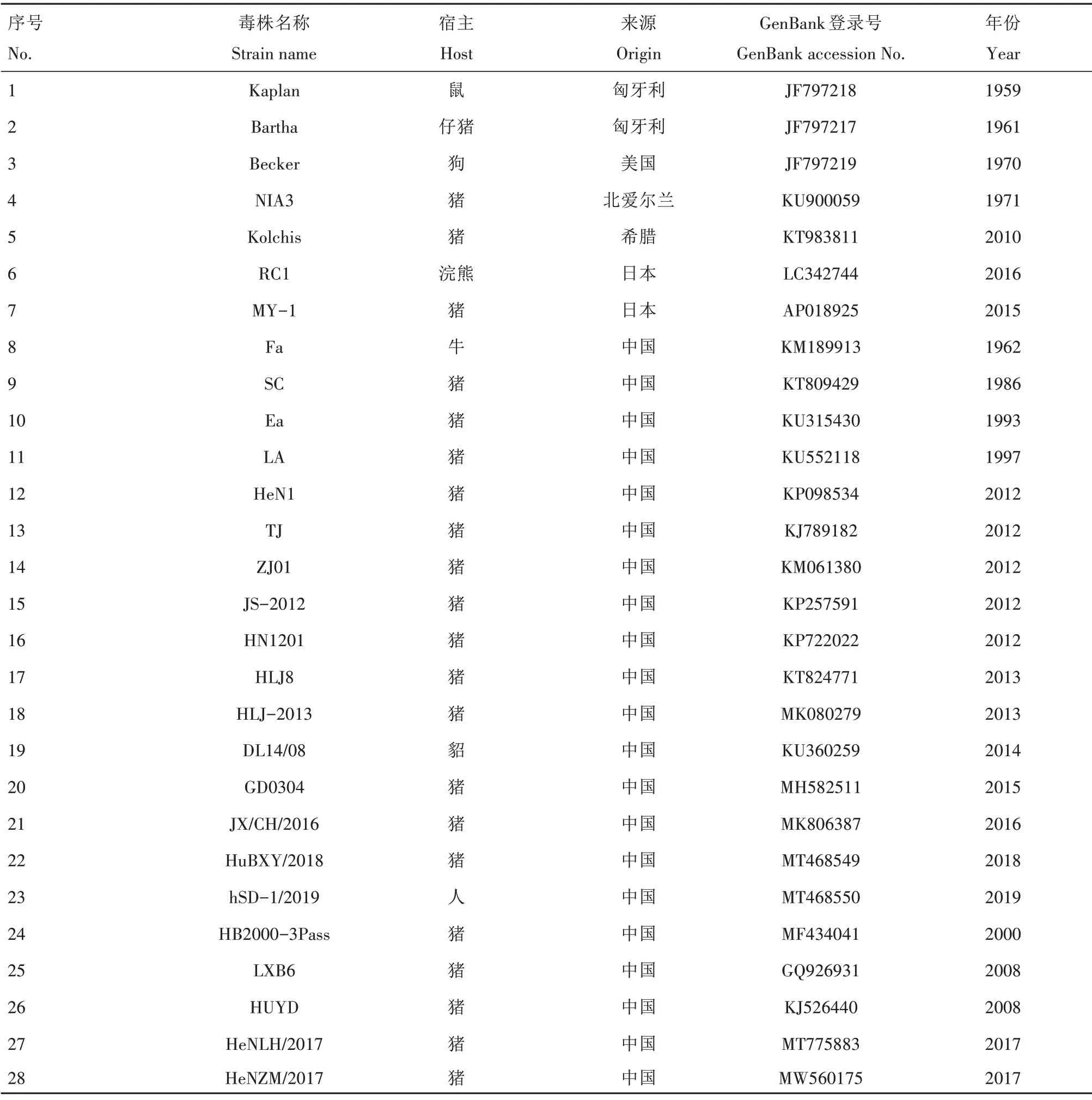
表1 毒株序列信息Tab.1 The strain sequence information in this study
2 结果与分析
2.1 PRV分离毒株基因组测序结果
按照基因组提取试剂盒(TaKaRa)操作说明提取细胞和PRV 的总DNA,采用Illumina NovaSeq二代测序方法(上海派森诺生物科技股份有限公司)完成了PRV 的全基因组序列测定,结果显示,HeNLH/2017 和HeNZM/2017 PRV 分离株基因组全长约为143 kb,GC 含量约为73.8%(表2),基因组结构与已报道的结果一致(图1),包含1 个UL区(Unique long region)和1 个US 区(Unique short region),以及位于US 区两侧的内部重复区(Internal repeat sequence,IR)和末端重 复区(Terminal repeat sequence,TR)。PRV 基因组共编码70 个开放阅读框(Open reading fram,ORF),其中,UL 区包含59 个基因,参与病毒DNA 复制以及病毒粒子的组装和成熟;US 区7 个基因,主要与病毒的感染和毒力有关;IR 区和TR 区则各包含2 个基因US1 和ICP4/IE180。2 株PRV 分离株的全基因组序列已上传至GenBank 数据库(MT775883 和MW560175)。
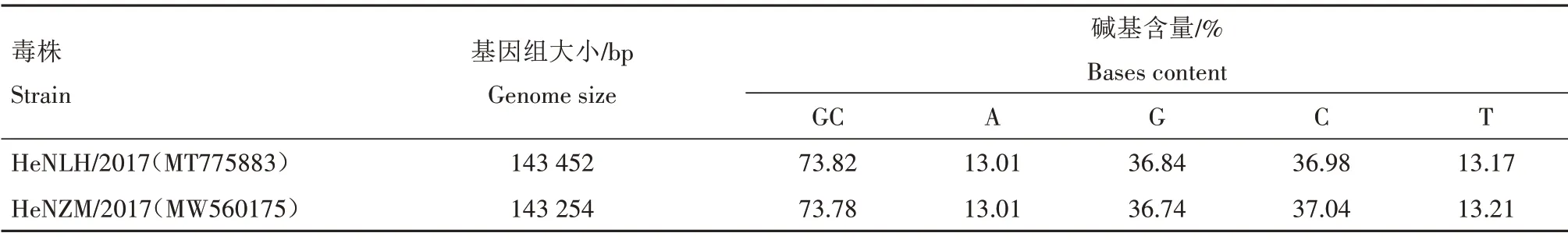
表2 2株PRV分离株基因组序列分析Tab.2 Genome sequence analysis of two PRV isolates
2.2 PRV分离毒株基因组同源性分析
PRV 基因组核苷酸同源性分析显示(图2),HeNLH/2017 和HeNZM/2017 PRV 分离株间的基因组核苷酸同源性为99.59%。我国流行毒株与欧美代表性毒株Bartha 株的遗传差异较大,为4.36%~4.72%;而我国流行毒株之间的同源性均较高,为98.31%~99.59%,其中2011 年以来流行的变异毒株与我国经典毒株Ea 和Fa 的同源性为98.31%~98.90%,遗传差异为1.10%~1.69%;变异毒株彼此之间的同源性为98.90%~99.59%,遗传差异为0.41%~1.10%。
2.3 PRV分离毒株遗传进化分析
分别基于PRV 全基因组序列和gC 基因序列构建了系统进化树(图3)。从图3 可以看出,PRV 毒株可以明显分为2 个基因型,基因1 型(Genotype 1)主要包括欧美地区的流行毒株,如Bartha、Kaplan 和Becker 等;基因2 型(Genotype 2)主要包括我国流行的毒株,又进一步分为基因2.1 亚型(Genotype 2.1)(以Ea 和Fa 株为代表)和基因2.2 亚型(Genotype 2.2)(以HeN1 和HN1201 株为代表)。本研究分离的HeNLH/2017 和HeNZM/2017 PRV 毒株均属于基因2.2亚型。
2.4 PRV分离毒株主要保护性抗原氨基酸变异分析
通过氨基酸序列比对,分析了PRV 不同毒株gB、gC和gD蛋白的氨基酸变异情况。从图4可以看出,与Bartha 株相比,2 株PRV 分离株以及我国其他PRV 流行毒株的gB 蛋白有36 个氨基酸的差异,包括75SPG77的缺失和94G 的插入,氨基酸差异达到3.93%;gC 蛋白有40 个氨基酸的差异,包括63AAASTPA69连续7个氨基酸的插入,氨基酸突变率达到了8.21%;gD 蛋白共有14 个氨基酸的差异,包括278S/RPRP281氨基酸的插入,氨基酸突变率约为3.47%。
与经典毒株Ea和Fa相比,2株PRV分离株以及我国2011 年以来流行的其他PRV 变异毒株氨基酸序列均非常保守,gB 蛋白仅有5 个氨基酸的差异,分别是T85A、R454K、H563K、T740A、V898A;gC 蛋白有3个氨基酸的变异,分别是T34N、E99K、G194E;gD 蛋白有278S/RP279的缺失和V338A位点的变异。
3 结论与讨论
PRV 基因组大小约140 kb,且具有高GC 含量(70%以上)和存在反复出现的重复序列等特征,这为PRV 全基因组序列的测定带来了困难[13]。近年来,随着二代测序技术的发展成熟和成本的降低,能够高效率地完成复杂基因组的测序工作。本研究利用Illumina NovaSeq 二代测序方法,成功获得了2 株PRV 变异毒株的全基因组序列。核苷酸同源性分析显示,HeNLH/2017 和HeNZM/2017 分离株与Bartha 株的核苷酸同源性为95.28%~95.37%;而与我国早期流行毒株Ea 和Fa 的同源性为98.70%~98.80%,与我国2011年以来新出现的变异毒株的同源性为98.90%~99.42%。说明分离毒株与变异毒株的核苷酸同源性更高。
根据地域分布和遗传差异,PRV 可以分为基因1 型和基因2 型[13-14]。基因1 型主要在欧美地区流行,并且大部分地区已经实现了该病的净化;而基因2 型则在亚洲主要是在中国流行,根据遗传和致病性差异,又进一步分为基因2.1 亚型和基因2.2 亚型[15]。基因2.1亚型即通常说的经典毒株,以我国早期分离株Ea 和Fa 为代表;基因2.2 亚型即通常所说的变异毒株,主要指2011 年以来引起我国PRV 再度流行的毒株。进化树分析显示,本研究分离的2株PRV 与我国流行的变异毒株处于同一进化分支,均属于基因2.2 亚型。此外,也有多个研究报道了PRV 不同毒株之间的基因重组现象,其中具有代表性的是SC 株和HLJ-2013 株,通过全基因组序列的分析,发现它们在Bartha 毒株与我国本土流行毒株之间发生了基因序列重组[15-18]。进化树分析也与这一发现一致,当以全基因组构建系统进化树时,SC株和HLJ-2013 株属于基因2 型;而以gC基因序列构建的系统进化树显示,SC 株和HLJ-2013 株同属于基因1型。
变异毒株的致病性显著高于经典毒株[19],且接种Bartha-K61 和Bucharest 疫苗后针对该类毒株的中和抗体水平较低[1,12];猪群接种Bartha-K61 疫苗后,可以针对我国早期流行毒株(Ea 和SC)提供良好的免疫保护,但对流行毒株(PRV-XT和JS-2012)仅能提供部分保护[12,20]。本研究采集病料的2 个猪场猪群均接种了Bartha-K61 疫苗,母猪每年接种4次,无明显临床症状;新生仔猪表现出神经症状、流涎等,多在48 h 内死亡。因此,有必要加紧研发针对我国PRV 新流行变异毒株的疫苗,从而更好地开展防控净化工作。
gB、gC和gD 蛋白是PRV 主要的保护性抗原,与不同流行毒株间的交叉保护和免疫逃逸紧密相关[9-12]。与Bartha疫苗毒株相比,我国PRV 流行毒株gB、gC 和gD 蛋白均广泛发生了氨基酸变异[21-22]。其中,gB 蛋白还包括特征性的75SPG77位的缺失和94G位的插入,gC 蛋白包括63AAASTPA69连续7 个氨基酸的插入,gD 蛋白包括278S/RPRP281位氨基酸的插入。此外,gB蛋白主要有3个抗原表位区域,分别是59—126 aa、214—279 aa 和540—734 aa[23],而gB 蛋白发生变异和插入的位置大部分位于59—126 aa和540—734 aa 抗原表位区,这在一定程度上解释了Bartha 疫苗对我国流行毒株保护效果不佳的原因。另外,需要注意的是,与早期PRV 毒株相比,我国流行的PRV 变异毒株较为保守(gD 蛋白具有278S/RP279的缺失),仅发生了个别氨基酸的变异,且这些发生变异的氨基酸与Bartha 株中对应的位点一致。在gB 蛋白发生变异的5 个氨基酸中,有4 个(T85A、H563K、T740A和V898A)与Bartha 株中一致;在gC 蛋白发生变异的3 个氨基酸中,有2个(T34N和E99K)与Bartha 株一致;gD 蛋白仅有的1 个氨基酸变异(V338A)也与Bartha 株一致。因此,变异毒株的免疫逃逸机制还有待进一步研究。
本研究测定分析了2 株PRV 的基因组遗传特征,并进一步分析了主要保护性抗原gB、gC 和gD 蛋白的氨基酸变异情况。我国流行毒株与欧美地区的毒株遗传关系较远,且主要保护性抗原均广泛发生了基因突变,并包含特征性的氨基酸插入或缺失序列。我国2011 年以来流行的变异毒株与早期流行毒株之间虽然存在一定的遗传差异,但整体同源性依然很高。
猜你喜欢
科学大观园(2022年2期)2022-01-23 11:05:15
天津市教科院学报(2021年5期)2021-11-10 07:32:40
生物学通报(2021年9期)2021-07-01 03:24:44
趣味(数学)(2020年4期)2020-07-27 01:44:16
支部建设(2020年15期)2020-07-08 12:34:32
江苏农业科学(2016年8期)2017-02-15 19:54:11
动物医学进展(2015年10期)2015-12-07 05:46:18
百科知识(2015年18期)2015-09-10 07:22:44
淮阴工学院学报(2014年5期)2014-09-10 09:15:56
特产研究(2014年4期)2014-04-10 12:54:12
