
层次聚类在进化树构建中的应用
2014-09-10 09:15李国宝
淮阴工学院学报 2014年5期
李国宝,业 宁
(南京林业大学 信息科学与技术学院,南京 210037)
0 引言
从达尔文时代开始,人类对生物起源的研究加以重视,各种生物进化学说相继被提出。由于生物进化历史是没有文字记载的,后人只能通过史前生物的化石等片面信息来尽可能准确的模拟生物进化的顺序,这就可能会形成错误的生物进化推断历史。随着20世纪中期生物遗传信息研究取得突破进展,人类通过生物的遗传物质来研究其进化历史成为可能。
现代生物学用生物进化树来描述生物之间进化关系,两种(或者多种)生物如果在同一层节点,则表明该组生物进化距离较近(即从同一祖先进化而来的可能性较大)[1];反之,表明这些生物之间的生物差异性较大。
生物进化树可以根据其是否按照进化距离构建来分类,这样就有基于进化距离构建的方法和基于统计特征或者生物离散特征构建的方法。基于进化距离的构建方法主要有最近邻法[2],UPGMA法等;基于统计方法的构建主要有最大似然法(Maximum likelihood)[3];基于生物离散特征的构建方法主要是最大简约法(Maximum Parsimony)[4]。
UPGMA方法是基于距离的进化树构建方法,该方法思想是:将两个进化距离最近的物种合成到一个复合物种组中,然后将新的距离矩阵中距离最小的两个物种再次合成一个复合物种组,如此反复,直到所有的物种都被聚为一棵进化树[5]。UPGMA方法的使用有一个前提,即假设一棵进化树中所有物种的进化速率[6]是相同的。
NJ(Neighbor Joining Method, NJ)方法是距离法建树中比较有实用价值的方法。与UPGMA相比,NJ方法不用假设进化树中所有物种的进化速率相同,因此在大多数情况下比较令人信服。该方法思想是:通过确定距离最近的成对分类物种组来使进化树的进化距离之和达到最小。在进行序列合并时,不仅要满足待合并序列进化距离的相近,同时也要求待合并的序列与其它序列的近似距离较远。本文实验一中生成的进化树将会与NJ方法生成的进化树进行比较。
ML(Maximum Likelihood)方法于1981年被提出,该方法构建思想基于统计学。在预先选择的进化模型下计算每一种进化树生成的可能性,选择最大可能性的进化树即为最大似然树。最大似然法在构建进化树的准确度方面很高,但是在处理大数据量时效率比较低,并且对模型的依赖比较严重。
MP(Maximum Parsimony)方法依据各个位置上由一条生物序列突变成另一条生物序列所需最小数量突变来进行比较分析和聚类树生成的,最终的进化树是基于整条序列所需的突变总数的。
1 研究思想
层次聚类的基本思想是:通过迭代分类,把相似的样本放在一层,直到样本都被归到某一层中[7]。具体的层次聚类算法分为两种,从顶层到底层的方法和从底层到顶层的方法。
从顶层到底层的算法思想是:(1)先把样本当作一个集合,从这个集合中取出2个(或者多个)最为相似的样本,形成一颗二叉(或者多叉)形状的树,此时,集合剩下的样本作为树根节点,2个(或者多个)相似的节点作为第一层叶子节点;(2)再从树根节点集合中选取一类相似的样本作为第二层叶子节点,继续扩展刚才生成的树;(3)重复步骤2,直到根节点集合中样本数为0;(4)按照2叉树的生成规则整理生成的进化树,保证结果是一棵2叉树。
从底层到顶层的层次聚类方法与前者描述的由顶层至底层算法相似,只是在进化树形成顺序上是从叶子节点到根节点。
先通过序列比对计算生物之间的进化距离,然后运用层次聚类方法[8]对生物样本进行分类,最终生成层次聚类结果以二叉树形式表现。与基于距离的其他方法如NJ、UPGMA等进化树构建方法以及基于统计的ML方法,基于生物表现特征的MP方法等生成的进化树比较后验证层次聚类方法的可行性,以及如何提高进化树的准确率。
2 序列比对计算生物进化距离
进化距离的计算是通过比较DNA序列得到的。序列的比对有两两比对和多重比对之分。比对算法有blast,clustal,fasta等,本实验采用的是多序列比对中的clustalw方法。
实验一:8个物种线粒体DNA距离矩阵
由于线粒体DNA变异速率很慢,它们以每一百万年百分之二点二的速度变异,因此选择线粒体DNA作为研究对象是比较合理的。
本实验将甘蓝型油菜种子萌发出现胚根第1天的幼苗转移至含有eBL(epi-Brassinolide)或BRZ的固体1/2 MS培养基上(方形皿)。eBL生理处理实验在光照条件下进行,6种处理浓度分别为0、1 × 10-9、1 × 10-8、1 × 10-7、1 × 10-6和 1 × 10-5 mol/L,处理4 d后观察不同浓度eBL对幼苗发育的影响。BRZ生理处理实验在黑暗下进行,分为0和1 × 10-6 mol/L 两个处理浓度,处理3 d后观察BRZ对幼苗黑暗条件下发育的影响。
序列来自GenBank,详细信息如表1所示。

表1 8 条线粒体 DNA 物种 GenBank 相关信息
由表1构建的距离矩阵见表2。
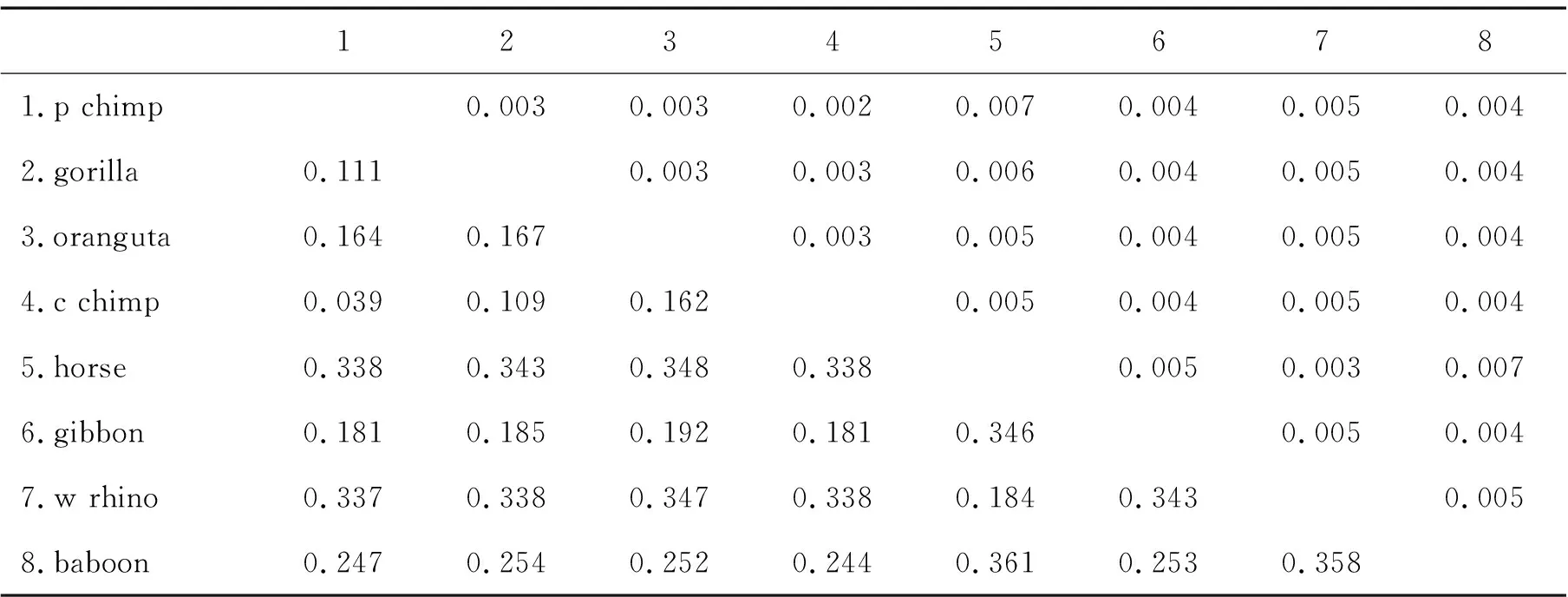
表2 8 个物种所对应的相似距离矩阵
实验二:十一种脊椎动物序列数据的实验
生物进化树的好坏,主要看与它与生物真实的进化历史差别多大。生物的真实进化史往往难以知晓,但是也有例外。表3给出的生物序列信息的真实进化树可以用古生物学和形态学方面的数据来构建[9]。11种脊椎动物线粒体全DNA信息如表3。

表3 11 种脊椎动物的名称及其线粒体全基因组 GenBank 编号
由表3构建的距离矩阵如表4所示。
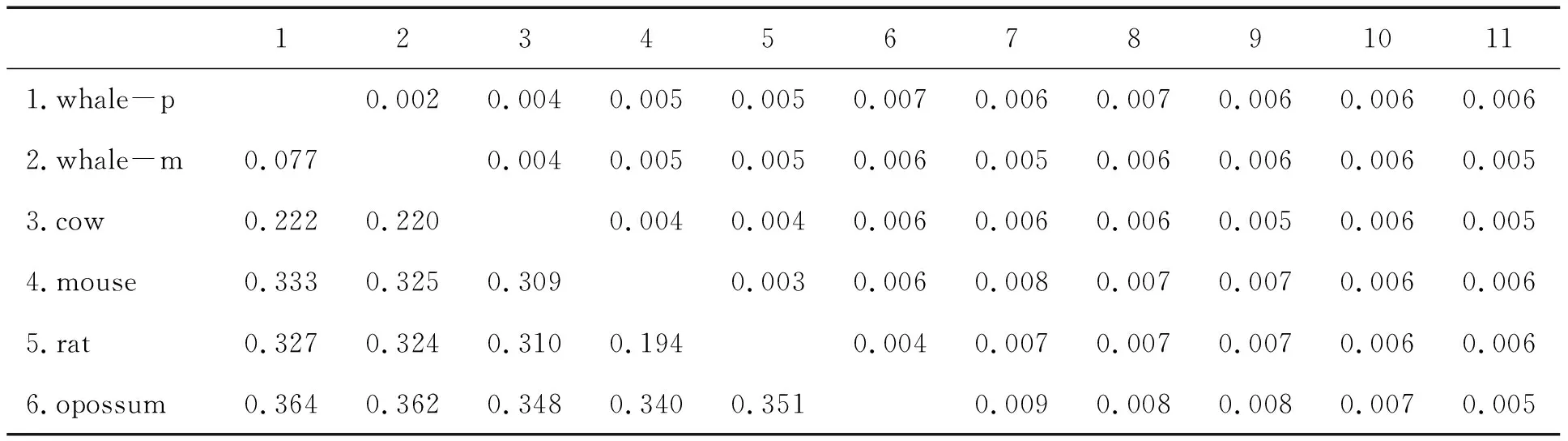
表4 11种脊椎动物线粒体DNA的距离矩阵
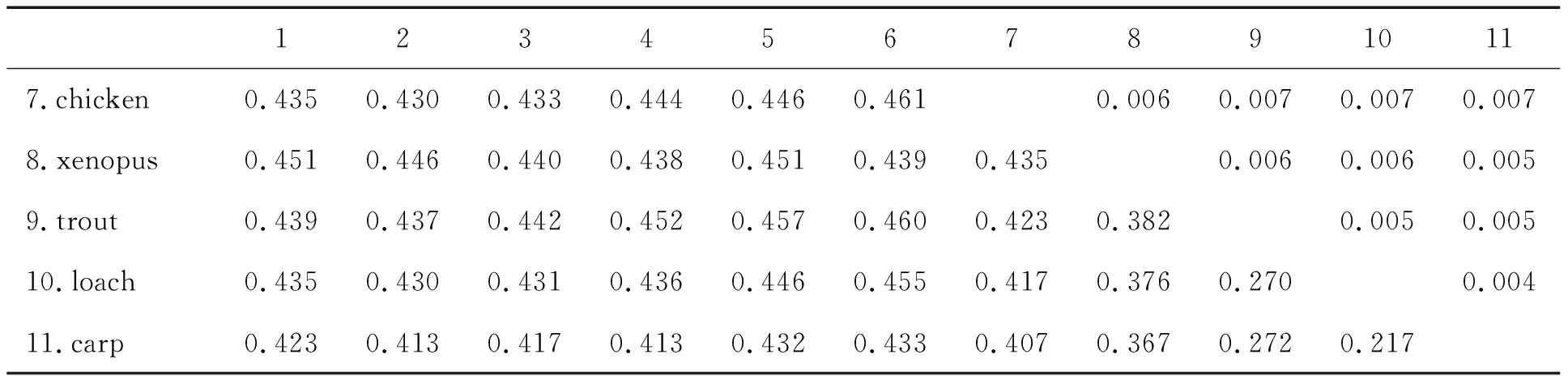
续表4 11种脊椎动物线粒体DNA的距离矩阵
3 层次聚类生成生物进化树
层次聚类的步骤:
(1)从n个序列样本中选择2个距离最小的放到集合s中,此时集合s和剩下的n-2个样本作为n-1个候选样本;
(2)从剩下的n-1个候选样本中选择2个距离最小的放到集合s1中,类似步骤1,此时n-2个候选样本;
(3)重复步骤2,直到候选样本树为0;
(4)此时n个序列样本层次聚类完成,通过层次号来逐层构建二叉树。
实验一通过层次聚类方法构建的进化树见图1。

图1 8个物种层次聚类结果
对生成的聚类结果统一注释后得到的进化树见图2。

图2 实验一的层次聚类生物进化树
实验二通过层次聚类的结果见图3。

图3 实验二11种脊椎动物线粒体
实验二层次聚类结果经过解释后的生物进化树见图4。
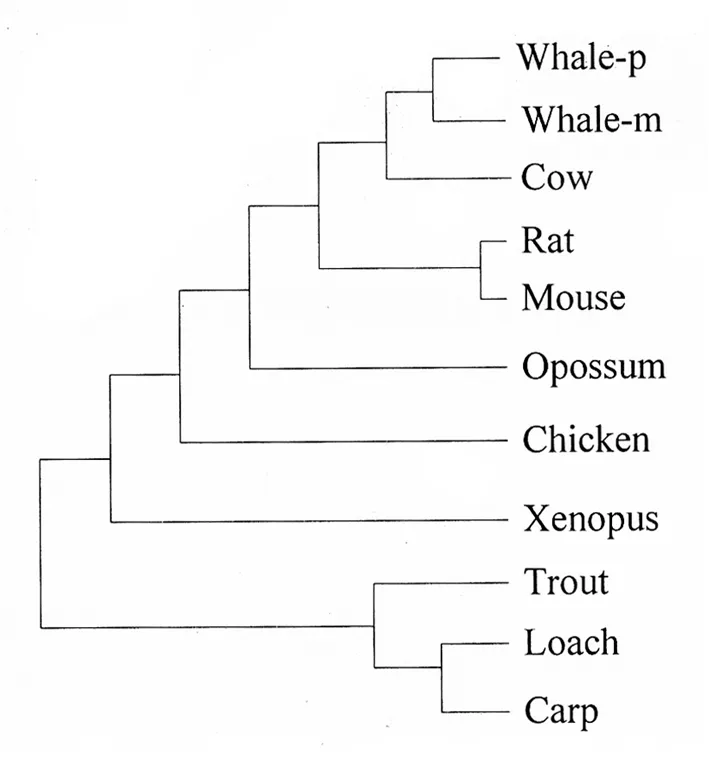
图4 实验二11种脊椎动物的生物进化树
4 对层次聚类生物进化树的评价
由于生物的真实进化历史无法得知,后人只能通过生物化石等信息来推断生物最有可能形成的进化历史。因此,对于构建的生物进化树,不存在唯一的评价标准。不过,可以在确保数据准确的前提下,对多种方法构建的进化树进行比较分析,如果多种方法构建的进化树拓扑结构一致,则可以侧面验证构建方法的有效性。
实验一中层次聚类构建的进化树与最大似然法、NJ法构建的进化树(见图5)结构完全一致,因此可以验证层次聚类在构建这8种生物线粒体DNA进化树中的有效性。

图5 最大似然法和最近邻法构建的8种生物进化树
实验二中层次聚类构建的11种脊椎动物的进化树和生物真实的进化树拓扑结构有细微差别,见图6。
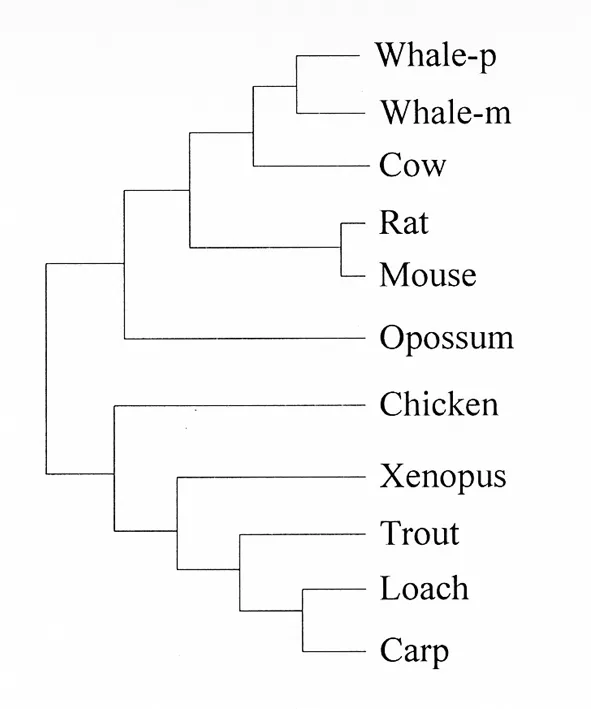
图6 11种脊椎动物的真实系统树
经过查阅相关资料,作者发现这种进化树结构差别源于所选取的序列长度较长。在计算相似距离时,序列越长,出现程序性错误的可能性就越大。
因此,本文的结论:在层次聚类产生进化树时,尽可能选取长度较短的能够代替生物进化信息的DNA序列,这样可以生成较准确的进化树。
参考文献:
[1] SNEATH P, SOKAL R. Numerical taxonomy—the principles and practice of numerical classification[M].San Francisco:W. H. Freeman and Company, 1973.
[2] SAITOU N, NEI M. The neighbor-joining method: a new method for reconstructing phylogenetic trees[J].Mol Biol Evol, 1987(4):406-425.
[3] Felsentein J. Evolutionary trees from DNA sequences: a Maximum likelihood approach[J].J Mol Evol, 1981,17:368-376.
[4] Kimura M. Evolutionary rate at the molecular level[J].Nature,1968,217:624-626.
[5] 刘金桂.分数阶超混沌系统的自适应函数投影同步[J].淮阴工学院学报,2012,21(1):1-4.
[6] Zhaxybayeva, O. and W.F. Doolittle, Lateral gene transfer[J].Current Biology, 2011,21(7):242-246.
[7] 孙亂,陆祖宏,谢建明.生物信息学基础[M].北京:清华大学出版社,2005.
[8] 丁淑妍.进化分析与结构预测中的若干问题研究[D].大连:大连理工大学,2012.
[9] 孙士保,秦克云.改进的k-平均聚类算法研究[J].计算机工程,2007,33(13):200-201.
猜你喜欢
天津市教科院学报(2021年5期)2021-11-10
海洋通报(2021年1期)2021-07-23
生物学通报(2021年9期)2021-07-01
生物学通报(2021年4期)2021-03-16
铁道通信信号(2019年6期)2019-10-08
中国医学创新(2017年7期)2017-03-31
雷达学报(2017年6期)2017-03-26
江苏农业科学(2016年8期)2017-02-15
电子设计工程(2015年6期)2015-02-27
癌变·畸变·突变(2014年1期)2014-03-01
