
川射干指纹图谱建立
2021-09-27 09:38:06刘晓凤姚周思敏曾尧王晨刘
中成药 2021年7期
刘晓凤 王 姚周思敏曾 尧王 晨刘 涛*
(1.西藏大学,西藏 拉萨850000; 2.四川天一学院,四川 绵竹618200; 3.成都大学,四川 成都 610106)
川射干属于鸢尾科植物鸢尾Iris tectorumMaxi.的干燥根茎[1],主产于广西、广东、四川等地区,为四川的道地药材,气微,味甘、苦,归肺经,具有清热解毒、消痰利咽的功效,主治咽喉肿痛、痰咳气喘。现代药理学研究表明,川射干在临床上还具有神经保护、抗肿瘤、抑菌等作用[2⁃5],具有良好的临床利用潜力。
2020 年版《中国药典》 一部收载川射干质量标准为用HPLC 法测定射干苷含量。据文献报道,川射干中还含有鸢尾甲苷A、鸢尾甲苷B、野鸢尾苷、鸢尾苷元、鸢尾甲黄素A、鸢尾甲黄素B 等多种异黄酮成分[6],具有保护心肌细胞、抗炎、抗肿瘤等功效[7⁃8],因此,选择单一的指标成分检测难以判别药材的优劣。本研究建立了川射干HPLC、UV、IR 指纹图谱,以期对该药材内在质量进行有效表征、综合评价和全面控制。
1 材料
P230Ⅱ型高效液相色谱仪(大连依利特分析仪器有限公司);TU⁃1810PC 型紫外可见分光光度计(北京普析通用仪器有限责任公司);Spectrum Two 型傅里叶变换红外光谱仪(美国Perkin Elmer 公司);FA2004 分析电子天平(上海良平仪器仪表有限公司);101⁃1⁃S 型电热鼓风干燥箱(成都雅源科技有限公司);PS⁃40 型超声波清洗器(深圳得康清洁设备有限公司)。
川射干均购自四川逢春制药有限公司,经成都大学刘涛研究员鉴定为鸢尾科植物鸢尾的干燥根茎,批号分别为170102、170108、170114、170123、170127、170206、170211、170221、170325、170331、170602、170608、170611、170615、170621、170708、170715、170723、170728、170813,分别编号为S1~S20,其中S3~S10、S15~S18 产自阿坝金川县,其他批次产自四川广元青川县。射干苷对照品,批号wkq16070103,纯度≥98%,购自四川省维克奇生物科技有限公司。甲醇和乙腈为色谱纯;其余试剂均为分析纯;水为纯净水。
2 方法与结果
2.1 HPLC 指纹图谱建立
2.1.1 色谱条件 Global Chromatography C18柱(4.6 mm×250 mm,5 μm);流动相乙腈(A)⁃0.1% 磷酸(B),梯度洗脱(0~15 min,20%~24%A;15~25 min,24%~26%A;25~36 min,26%~30% A;36~50 min,30%~32% A;50~70 min,32%~20% A);体积流量1.0 mL/min;柱温30 ℃;检测波长280 nm;进样量10 μL。
2.1.2 对照品溶液制备 取射干苷对照品适量,精密称定,加70%乙醇制成质量浓度为43 μg/mL 的溶液,即得。
2.1.3 供试品溶液制备 取川射干(过3 号筛)约0.5 g,精密称定,具塞锥形瓶中,精密加入25 mL 甲醇,密塞,称定质量,超声(240 W、40 kHz)处理30 min,放冷,再称定质量,补足,摇匀,滤过,取续滤液l mL,置5 mL 量瓶中,加甲醇稀释至刻度,摇匀,即得。
2.1.4 方法学考察
2.1.4.1 精密度试验 取同一供试品溶液,在“2.1.1”项色谱条件下进样6 次,测得各共有峰相对保留时间RSD均小于0.5%,各共有峰相对峰面积RSD 均小于3.0%,表明仪器精密度良好。
2.1.4.2 重复性试验 取同一批样品,按“2.1.2”项下方法平行制备供试品溶液6 份,在“2.1.1”项色谱条件下进样,测得各共有峰相对保留时间RSD 均小于0.5%,相对峰面积RSD 均小于3.5%,表明该方法重复性良好。
2.1.4.3 稳定性试验 取同一供试品溶液,于0、2、4、6、8、12 h 在“2.1.1”项色谱条件下进样,测得各共有峰相对保留时间RSD 均小于0.3%,相对峰面积RSD 均小于4.5%,表明溶液在12 h 内稳定性良好。
2.1.5 图谱采集 取10 批样品,按“2.1.2”项下方法平行制备供试品溶液,在“2.1.1”项色谱条件下进样,结果见图1。
2.1.6 图谱生成 采用中国药典委员会“中药色谱指纹图谱相似度评价系统(2004A 版)”,对10 批样品的指纹图谱进行分析,生成对照指纹图谱。取射干苷对照品溶液,在“2.1.1”项色谱条件下测定,共确定7 个共有峰,1 号峰为射干苷,结果见图1。
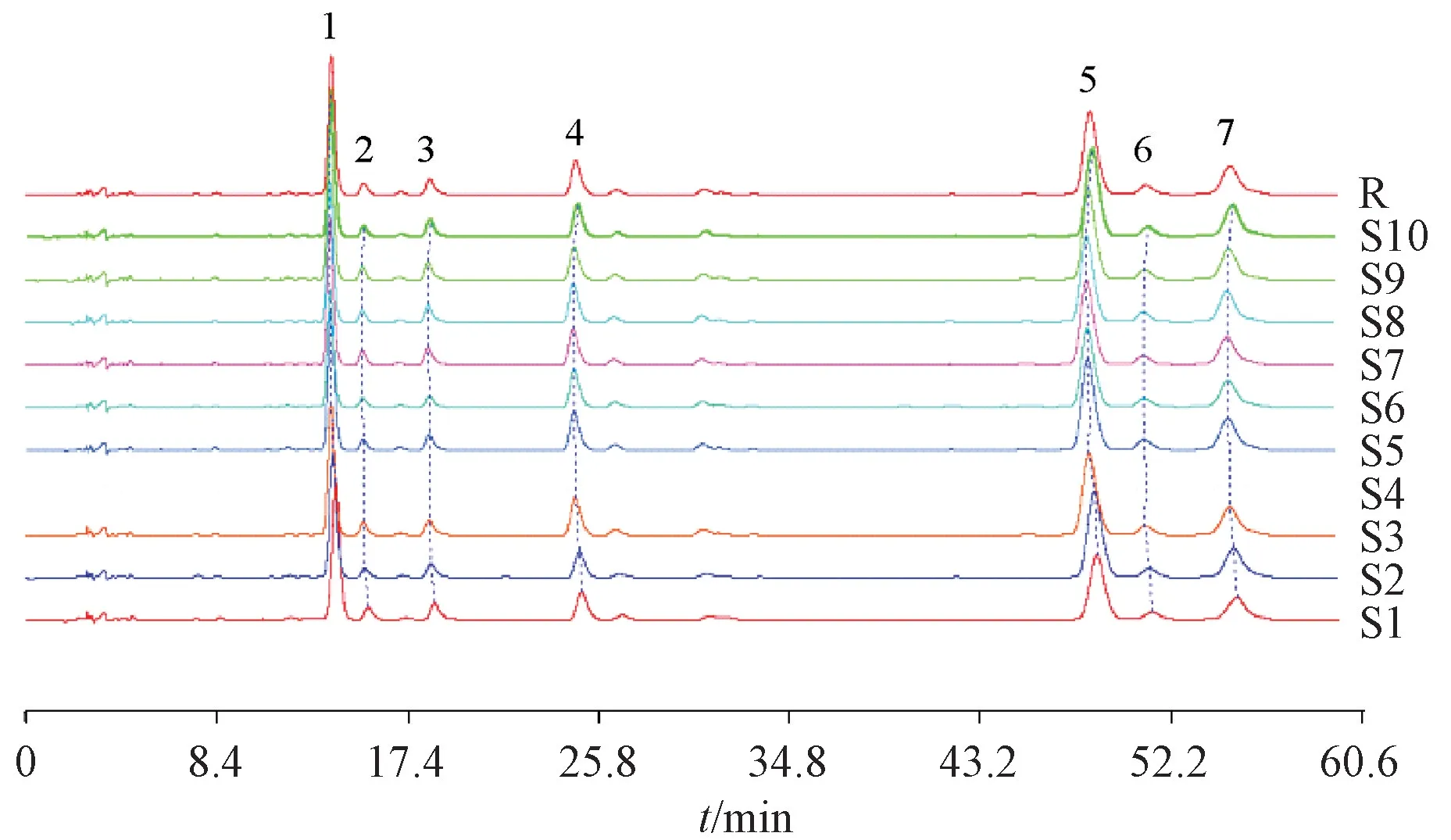
图1 10 批样品HPLC 指纹图谱
2.1.7 共有峰相对保留时间及相对峰面积 在HPLC 指纹图谱中,1 号峰在各批样品中均有出现,分离度良好,峰面积大,故选作参照峰,将其保留时间和峰面积记为1.000 0,其他共有峰相对保留时间、相对峰面积分别见表1~2。

表1 10 批样品共有峰的相对保留时间

表2 10 批样品共有峰的相对峰面积
2.1.8 相似度评价 采用中国药典委员会“中药色谱指纹图谱相似度评价系统(2004A 版)”,将10 批样品的指纹图谱与对照指纹图谱进行相似度分析,结果分别为0.990、0.997、0.999、0.999、1.000、0.998、0.999、1.000、0.997、0.999,平均值0.998。将相似 度平均 值80%~120%作为相似度变异可接受范围,拟规定两者相似度应大于0.798。
2.1.9 验证试验 另取10 批样品,按“2.1.2”项下方法制备供试品溶液,在“2.1.1”项色谱条件下进样。运用中国药典委员会“中药色谱指纹图谱相似度评价系统(2004A 版)”,将10 批样品的指纹图谱与对照指纹图谱进行相似度分析,结果分别为0.995、0.997、1.000、0.999、1.000、0.999、0.999、1.000、0.999、1.000,符合上述规定,相对保留时间、相对峰面积分别见表3~4。

表3 10 批样品(验证)共有峰的相对保留时间
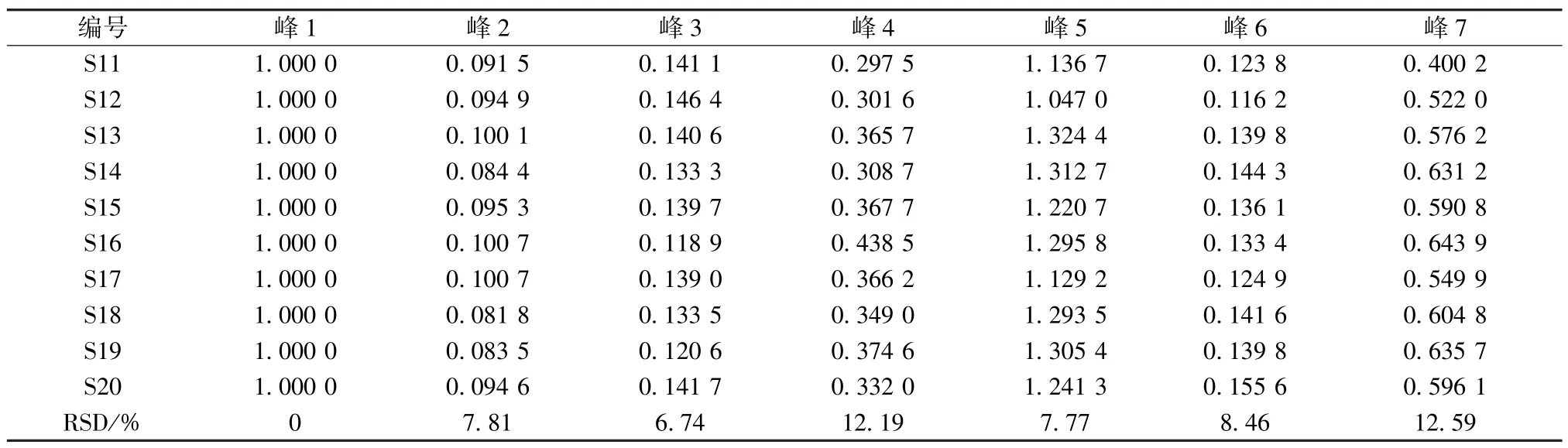
表4 10 批样品(验证)共有峰的相对峰面积
2.2 紫外指纹图谱建立
2.2.1 光谱扫描条件 以95%乙醇为空白调整基线,扣除基底影响。光谱扫描条件为光度模式,光谱带宽2.00 nm,扫描范围200.00~800.00 nm,扫描间隔1.0 nm,快速扫描。
由于川射干主要成分为黄酮类,其吸光度在紫外光区,故对照指纹图谱及相似度均以200~400 nm 处的吸光度为标准进行建立并计算。
2.2.2 供试品溶液制备 取药粉约0.5 g,精密称定,具塞锥形瓶中,精密加入25 mL 95%乙醇,密塞,称定质量,超声(240 W,40 kHz)处理30 min,放冷,再次称定质量,补足,摇匀,滤过(续滤液稀释312.5 倍时,图谱吸光度均处于0.2~0.8 之间),摇匀,即得。
2.2.3 方法学考察
2.2.3.1 精密度考察 取同一供试品溶液,在“2.2.1”项条件下连续扫描6 次,得210、240、266 nm 处紫外吸光度RSD 分别为0.08%、0.52%、0.30%,表明仪器精密度良好。
2.2.3.2 重复性考察 取同一批样品,按“2.2.2”项下方法平行制备供试品溶液6 份,在“2.2.1”项条件下进行扫描,测得210、240、266 nm 处紫外吸光度RSD 分别为2.95%、2.74%、3.20%,表明该方法重复性良好。
2.2.3.3 稳定性考察 取同一供试品溶液,于0、0.5、1、1.5、2、3 h 在“2.2.1”项条件下进行扫描,测得210、240、266 nm 处紫外吸光度RSD 分别为2.06%、2.02%、3.07%,表明溶液在3 h 内稳定性良好。
2.2.4 图谱采集 取10 批样品,按“2.2.2”项下方法制备供试品溶液,在“2.2.1”项条件下测定,紫外光谱叠加图见图2。

图2 10 批样品紫外指纹图谱
2.2.5 图谱生成 将“2.2.4”项下紫外图谱各波长下的吸光度取平均值,并以其建立对照指纹图谱[9],见图3。

图3 紫外对照指纹图谱
2.2.6 相似度分析 将10 批样品的紫外指纹图谱与对照紫外指纹图谱经IBM SPSS statistics 软件中双变量相关“Kendall 的tau⁃b”分析计算相似度[10]。结果,相似度分别为0.991、0.995、0.993、0.996、0.998、0.996、0.990、0.996、0.994、0.995,平均值 为0.994,将其平 均值的80%~120%作为变异可接受范围,拟规定两者相似度应大于0.795。
2.2.7 验证试验 另取10 批样品,按“2.2.2”项下方法制备供试品溶液,在“2.2.1”项条件下扫描,所得紫外图谱与对照指纹图谱各波长下吸光度经数据处理软件IBM SPSS statistics 中双变量相关“Kendall 的tau⁃b 分析”计算相似 度。结果,相似度 分别为0.960、0.961、0.959、0.956、0.956、0.960、0.959、0.960、0.957、0.957、0.957,符合上述规定。
2.3 红外指纹图谱建立
2.3.1 供试品溶液制备 取药材约0.5 g,精密称定,具塞锥形瓶中,精密加入25 mL 95%乙醇,密塞,称定质量,超声(240 W、40 kHz)处理30 min,放冷,补足减失质量,摇匀,滤过。精密移取0.2 mL 续滤液于玛瑙研钵中,吹干溶剂,与约0.1 g 干燥KBr 粉末研磨均匀,于14 MPa真空环境中压片1.5 min,即得。
2.3.2 光谱扫描条件 取按“2.3.1”项下方法制备的供试品薄片,放入傅里叶变换红外光谱仪中扫描,并扣除空气干扰,扫描范围4 000~400 cm-1。
2.3.3 方法学考察
2.3.3.1 精密度试验 取药材粉末约0.5 g,按“2.3.1”项下方法制备供试品溶液,在“2.3.2”项条件下连续扫描6 次。取图谱中吸收强度最强的10 个峰作为特征峰,测得特征峰波数RSD[11]均小于0.02%,表明仪器精密度良好。
2.3.3.2 重复性试验 取同一批样品,按“2.3.1”项下方法平行制备供试品溶液6 份,在“2.3.2”项条件下扫描,测得特征峰波数RSD 均小于0.25%,表明该方法重复性良好。
2.3.3.3 稳定性试验 取药材粉末约0.5 g,按“2.3.1”项下方法制备供试品溶液,在“2.3.2”项条件下于0、0.5、1、1.5、2、3 h 扫描,测得特征峰波数RSD 均小于0.02%,表明溶液在3 h 内稳定性良好。
2.3.4 图谱生成 将20 批样品按“2.3.1”项下方法制备供试品溶液,在“2.3.2”项条件下扫描,红外光谱叠加图见图4,各批次吸收峰的波数见表5。
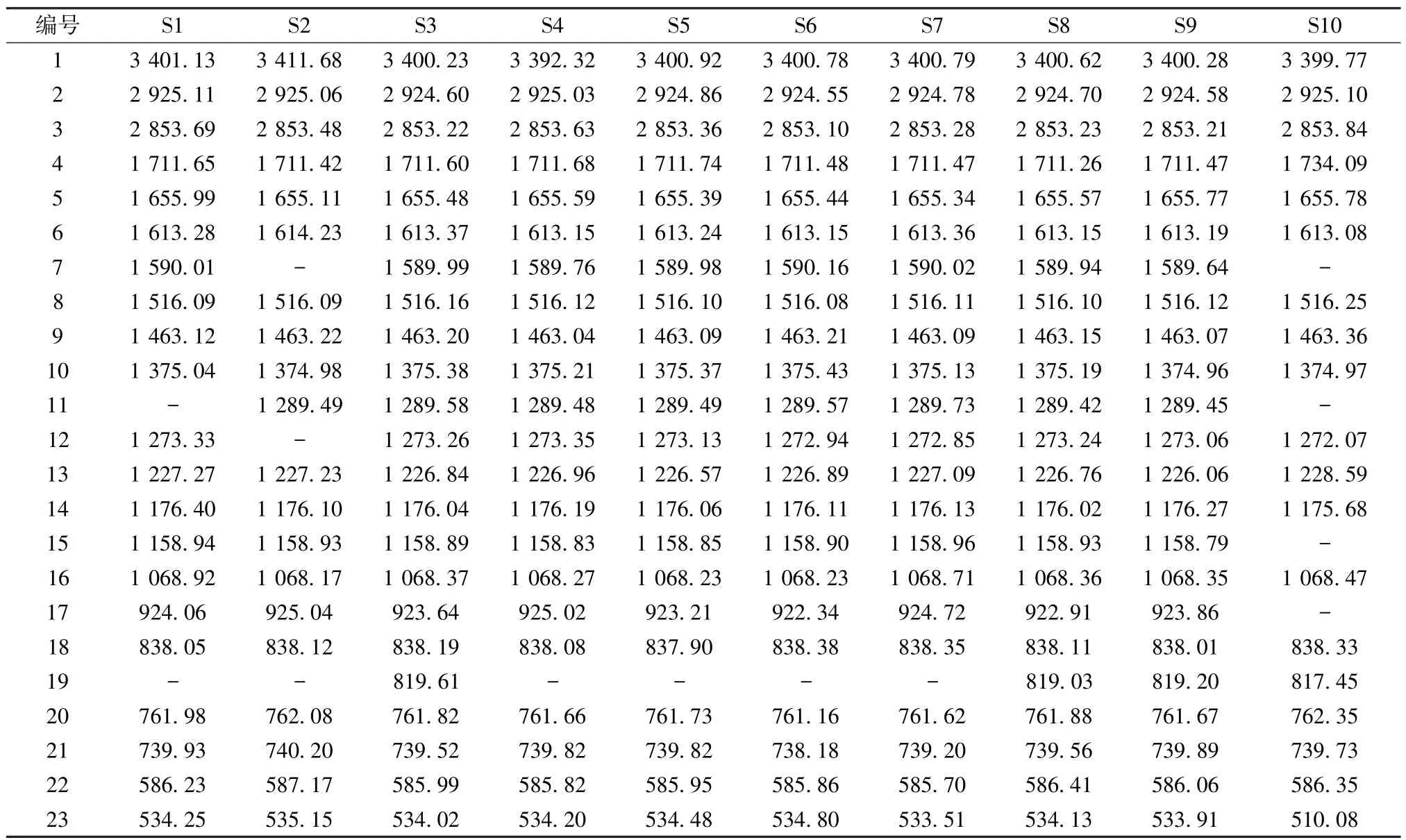
表5 20 批样品红外吸收峰波数(cm-1)

续表5

图4 20 批样品红外指纹图谱
2.3.5 共有峰确定 若组内吸收峰的波数最大差异显著小于与其相邻组之间的平均波数差,就确定该组峰是一组共有峰,反之为变异峰[12⁃13]。
2.3.6 双指标序列分析法鉴定指标 双指标序列为以某一批次样品作参考,计算其他样品红外指纹图谱的共有峰率和变异峰率,并依次排列而成的序列。
2.3.7 双指标序列分析 对20 批样品进行双指标序列分析,结果表明,红外指纹图谱共有峰率65.22%~100.00%,变异峰率0~27.78%。其中,产自阿坝金川县的样品之间共有峰率78.26%~100.00%,变异峰率0~22.22%;产自四川广元青川县的样品间共有峰率73.91%~100.00%,变异峰率0~16.67%,表明同一产地样品之间共有峰率高,变异峰率低。
3 讨论
现有对川射干在质量控制方面的研究,多采用HPLC方法测定一种或者多种有效成分含量,方法较为繁琐,且中药化学成分复杂,测定单一指标成分并不能完全代表该药材的品质。而指纹图谱就是研究以反映中药的整体化学特征为立论依据,对药品质量进行整体评价的方法。HPLC指纹图谱是在确定中药化学成分的基础上对中药进行整体控制,广泛应用于中药材及中成药的真伪优劣鉴别[14⁃17]。紫外指纹图谱可反映中药中具有不饱和化学键及其共轭体系基本物质的吸收情况,并以此控制中药质量[18⁃19]。红外指纹图谱可反映中药中多种化合物成分饱和键的吸收,并确定该中药中的各种化学基团,以此控制中药质量[11⁃13]。
本实验建立了HPLC、UV、IR 指纹图谱,其中HPLC、UV、IR 指纹图谱特征均有一定差异,故将多种分析方法联合或许能够减少单一检测分析方式在某些方面的不足而造成的误差,使川射干药材能得到简便、可靠、完整、健全的质量控制。但此方法不能确定药材中的化学成分及药理活性的关系,有待进一步研究。
猜你喜欢
小哥白尼(趣味科学)(2021年11期)2021-02-28 08:35:00
少先队活动(2020年12期)2021-01-14 01:47:40
小天使·一年级语数英综合(2020年10期)2020-12-16 02:57:11
天然产物研究与开发(2018年8期)2018-09-10 05:48:28
基层中医药(2018年5期)2018-08-31 02:35:54
天然产物研究与开发(2018年2期)2018-04-04 02:01:24
中成药(2017年3期)2017-05-17 06:09:01
领导科学论坛(2016年9期)2016-06-05 14:59:58
自动化学报(2016年8期)2016-04-16 03:39:00
青少年科技博览(中学版)(2015年7期)2015-08-12 18:50:24
