
CXCR3及其配体在肿瘤免疫治疗中的研究进展①
2021-09-25 03:19:42肖文璐蒋敬庭卢斌峰
中国免疫学杂志 2021年16期
肖文璐 蒋敬庭 卢斌峰
(苏州大学附属第三医院肿瘤生物诊疗中心,江苏省肿瘤免疫治疗工程技术研究中心,苏州大学细胞治疗研究院,常州213003)
趋化因子是一类与G蛋白耦联受体亚群相互作用的小分子蛋白,根据N端两个保守半胱氨酸残基的位置,可分为两个主要亚家族(CX3C和CXC)和两个次要亚家族(CC和C)[1]。趋化因子能够与表达相应受体的靶细胞相结合并介导其功能[2]。趋化因子受体CXCR3由3种IFN-γ诱导型配体CXCL9、CX‐CL10和CXCL11激活[3]。CXCR3活化后优先在Th1型CD4+T细胞、效应CD8+T细胞、NK细胞以及NKT细胞上高表达。CXCR3在效应T细胞向炎症和肿瘤部位的迁移中起关键作用[4]。
根据是否具有ELR(谷氨酸-亮氨酸-精氨酸)基序,将CXC趋化因子分为两种形式:具有ELR基序者可促进嗜中性粒细胞迁移和血管生成,不具有ELR基序者能促进淋巴细胞迁移并抑制血管生成[5]。CXCL9,10,11是ELR阴性的CXC趋化因子,可通过抑制血管生成,发挥抗肿瘤作用。然而,也有研究发现CXCL9,10,11增加肿瘤增殖和转移,可能是由于配体对CXCR3变体(CXCR3-A,CXCR3-B和CXCR3-alt)产生的不同影响[6]。CXCL9,10,11/CXCR3通路主要调节免疫细胞的活化、分化和迁移,通过募集免疫细胞,如细胞毒性淋巴细胞(cyto‐toxic lymphocytes,CTL)、NK细胞、NKT细胞和巨噬细胞等发挥免疫应答效应。
1 CXCR3及其配体
CXCR3,也称为G蛋白偶联受体9(GPR9)或CD183,是一种7跨膜结构域G蛋白偶联受体。人类趋化因子受体CXCR3存在3种剪接变体:CXCR3-A,CXCR3-B和一个截短的变体CXCR3-alt。CX‐CR3-A和CXCR3-B的 配 体 为CXCL9、CXCL10和CXCL11,而CXCR3-alt仅 结 合CXCL11[7]。CXCR3在CD4+Th1细胞、CD8+细胞毒性T细胞以及NK和NKT细胞上表达,但在初始T细胞上不表达。CX‐CL9和CXCL10均能增强Th1细胞的效应功能,但CXCL11结合在CXCR3受体上的不同位点,并介导相反的功能,促进分化Foxp3−调节性T细胞(T-regu‐latory 1,Tr1)的表达,抑制效应T细胞的功能[8-9]。
趋化因子CXCL9,亦称IFN-γ诱导的单核因子(MIG),在免疫细胞的趋化中发挥重要作用,可由多种细胞分泌,包括免疫细胞(T淋巴细胞、NK细胞、树突状细胞、巨噬细胞和嗜酸性粒细胞等)和非免疫细胞(内皮细胞,肿瘤细胞和成纤维细胞等)[10]。CXCL9主要介导淋巴细胞浸润至局灶性部位并抑制肿瘤生长。
趋化因子CXCL10,亦称IFN-γ诱导蛋白10(IP-10)。人类CXCL10基因最初发现是在淋巴瘤细胞系(U937)中由IFN-γ诱导的早期反应基因。CX‐CL10在胸腺、脾脏和淋巴结基质中以低水平表达,可通过IFN-α、IFN-β、IFN-γ或LPS刺激,诱导其在多种细胞高表达,包括内皮细胞、成纤维细胞、单核细胞和中性粒细胞等,是活化T细胞的化学诱导物[11]。CXCL10除诱导效应Th1细胞外,还可将CX‐CR3+CD8+T细胞募集到肿瘤部位,并通过这些细胞诱导颗粒酶-b的产生,从而增强抗肿瘤效应[12]。
趋化因子CXCL11,亦称干扰素诱导的T细胞α化学引诱物(I-TAC)或IFN-γ诱导蛋白9(IP-9)。CXCL11是与CXCR3结合亲和力最高的配体,这种相互作用促进嗜酸性粒细胞从骨髓中释放并进入外周组织[1]。此外,CXCL11可与CXCR7结合,这与细胞的侵袭性相关并减少肿瘤细胞的凋亡[13]。
2 CXCL9,10,11/CXCR3通路在肿瘤中的作用
CXCL9,10,11主要由单核细胞、内皮细胞、成纤维细胞和肿瘤细胞分泌产生。CXCL9,10,11/CXCR3通路有两种作用方式:免疫激活的分泌信号和肿瘤细胞衍生的增殖转移信号[14]。免疫激活的分泌信号主要作用于免疫细胞的迁移、分化和激活,通过该通路募集CTL、NK细胞、NKT细胞和巨噬细胞发生免疫反应性,介导Th1细胞极化,通过IFN-γ反应而激活免疫细胞。对于肿瘤细胞衍生信号,肿瘤衍生的配体主要通过CXCR3-A使肿瘤细胞具有转移倾向,并且肿瘤衍生的趋化因子促进Th2细胞、调节性T细胞(regulatory T cells,Tregs)和骨髓来源的抑制性细胞(myeloid-derived suppressor cell,MDSC)的募集,产生促肿瘤作用(见图1)。
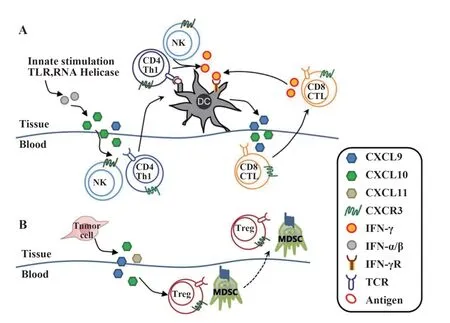
图1 CXCL9,10,11/CXCR3通路在肿瘤微环境中的作用[14-15]Fig.1 CXCL9,10,11/CXCR3 pathway in tumor micro⁃environment[14-15]
2.1 抗肿瘤作用 CXCL9和CXCL10是许多免疫反应的关键组成部分,通过与CXCR3结合激活NK和Th1细胞,并诱导其进入炎症部位。ADDISON等[16]发现人类早期非小细胞肺癌中高表达ELR−CX‐CL9,重组人细胞因子(recombinant human cytokine CXCL9,rhCXCL9)或CXCL9的基因转移均可抑制肿瘤衍生的血管生成,抑制肿瘤的生长和转移。在乳腺癌小鼠模型中发现,肿瘤细胞表达的CXCL9通过募集宿主NK细胞和大量CXCR3+CD4+及CX‐CR3+CD8+宿主T细胞抑制局部肿瘤生长和转移[17]。CXCL9和CXCL10的高表达与CD8+T细胞的高密度显著相关,能够募集记忆CD8+T细胞和巨噬细胞。在Treg细胞耗竭的肿瘤中,CXCL9和CXCL10选择性增加,CXCR3+T细胞募集,这表明靶向Treg细胞并上调CXCL9和CXCL10的表达可作为一种潜在的免疫疗法[18]。
CXCL10对肿瘤发生具有双重作用,这取决于相应CXCR3受体的剪接变体。CXCR3-B具有生长抑制特性,而CXCR3-A促进细胞增殖[19]。CXCL10与肿瘤细胞表面的CXCR3-B受体结合,直接发挥抗增殖作用,缩短肿瘤细胞的生存期;与CXCR3-A受体结合,减弱FAS(CD95)介导的细胞凋亡作用[20]。CXCL10是有效的血管生成阻断剂,与CXCR3受体结合后,抑制肿瘤血管生成从而降低肿瘤内血管密度,增加肿瘤细胞凋亡使肿瘤组织坏死[21]。CXCL10可招募多种免疫细胞直接杀伤肿瘤细胞,发挥抗肿瘤作用[12]。另外,CXCL10还可干扰致癌因子的表达,促使肿瘤细胞凋亡[22]。
2.2 促肿瘤作用 CXCL9与受体亚型CXCR3-A结合,激活rhCXCL9诱导的p-ERK1/2-MMP2/MMP9通路,增强CD133+肝细胞癌的迁移和侵袭能力[23]。EJAEIDI等[24]发现在转移性乳腺癌患者血清中,CX‐CL9、CXCL10和CXCL11的表达水平明显升高。血清中的这3种趋化因子,通过激活存活蛋白、β-链蛋白、丝裂原活化的蛋白激酶磷酸酶1(MKP-1)和基质金属蛋白酶1(MMP-1),在乳腺癌的发展中发挥重要作用。此外,抑制CXCL9的表达可促进细胞内肌动蛋白聚合,细胞黏附和细胞存活,还可增加细胞内的钙浓度,诱导黑色素瘤细胞的迁移[25]。
肿瘤细胞分泌CXCL10,趋化CXCR3+CD4/CD8/Treg细胞进入肿瘤组织,CXCL10的促进或抑制作用可能取决于CXCR3+CD8+T细胞与CX‐CR3+Treg细胞间的平衡。CXCL10变体拮抗免疫细胞募集,使早期T细胞募集功能受损,增加Treg细胞的渗透比率。急性期肝移植后增强的CXCL10/CX‐CR3信号直接诱导Treg细胞的动员和募集,从而促进肝细胞癌的生长和移植后的复发[26]。因此,高表达CXCL10及CXCR3的某些肿瘤细胞具有更强的转移和侵袭能力。
3 CXCL9,10,11/CXCR3通路作为肿瘤免疫治疗的靶点
CXCL9,10,11/CXCR3途径通过激活免疫活化分泌信号,抑制肿瘤细胞衍生信号作为肿瘤治疗的新靶点(见表1)。将Th1细胞、CTL、NK细胞、NKT细胞和M1巨噬细胞募集到肿瘤部位的配体可作为有效的抗肿瘤策略。携带CXCL9的质粒载体联合顺铂治疗可控制结肠癌和肺癌的发展,增强CTL的激活[27]。在肾细胞癌模型中,肿瘤内CXCL9和系统性IL-2通过肿瘤内浸润的CXCR3+单核细胞抑制肿瘤生长和血管生成[28]。在骨髓瘤小鼠模型中,CX‐CL10-Ig融合蛋白具有更长的半衰期,并保持重组蛋白的特征,可诱导CTL和NK细胞浸润到肿瘤部位[29]。另外,一种新的CXCL10-EGFRvIII融合蛋白(IP10-scFv)在神经胶质瘤小鼠模型中成功诱导了肿瘤浸润淋巴细胞,延长小鼠生存期[30]。由于CX‐CL11可诱导Treg细胞迁移或促进Tr1和Th2细胞的极化,因此其能否作为肿瘤治疗的靶点可能存在争议。CXCR3的拮抗剂AMG487在体外抑制结肠癌和骨肉瘤细胞的生长,在体内模型中抑制肺转移,但AMG487不能抑制肝转移和转移瘤的生长[31-32]。AMG487靶向CXCR3的所有变体,抗CXCR3可通过旁分泌CXCL9,10,11/CXCR3途径抑制肿瘤转移,还可抑制抗肿瘤宿主反应。
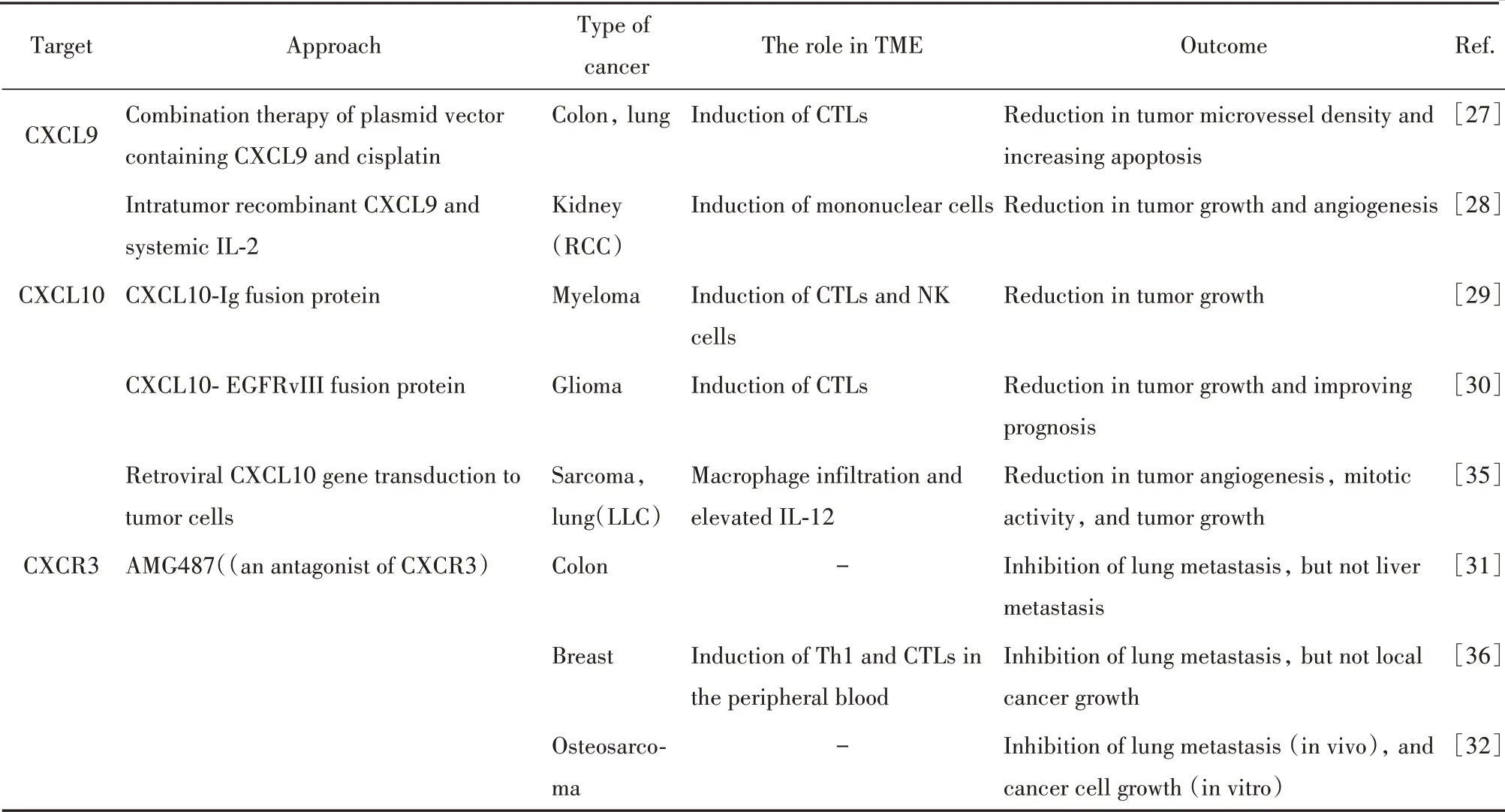
表1 基于CXCL9,10,11/CXCR3通路的肿瘤免疫治疗研究Tab.1 Tumor immunotherapy based on CXCL9,10,11/CXCR3 pathway
CXCL9,10,11/CXCR3通路与PD-1/PD-L1之间的关系是一个重要的研究领域,将该途径与其他免疫疗法相结合,通过多种机制改善对肿瘤发展的抑制作用,提高肿瘤免疫治疗的疗效。CHOW等[33]揭示了抗PD-1免疫疗法的成功应用需要CD8+T细胞表达CXCR3,以及CD103+DC产生CXCL9。在抗PD-1治疗的荷瘤小鼠中,CXCR3及其配体在产生CD8+T细胞反应中至关重要,肿瘤内CD103+DC细胞产生CXCL9促进抗PD-1诱导的抗肿瘤反应,表明CXCR3促进了肿瘤微环境中DC与T细胞之间的相互作用[33]。PD-1和CTLA-4免疫检查点双重阻断后,CXCL9和CXCL10的表达显著上调,CD8+T细胞浸润和治疗效果均依赖CXCR3。在荷瘤小鼠模型和经免疫检查点阻断治疗的患者中发现,巨噬细胞是这两种趋化因子的主要来源,其消耗可降低CD8+T细胞的浸润和免疫检查点双重阻断的疗效[34]。
4 展望
目前人们已经了解到CXCR3趋化因子系统在炎症过程中的作用,既增加CD4+和CD8+T淋巴细胞募集,驱动炎症反应,也可诱导和募集Th1型特异性调节T细胞发挥抑制作用。然而CXCR3如何调节这些关键反应之间的平衡有待进一步研究。CX‐CR3及其配体在血管生成、肿瘤生长和转移中具有重要作用,但对于肿瘤组织中趋化因子的表达和定位,以及CXCL9,10,11/CXCR3通路在淋巴和外周组织中T细胞迁移的作用需要进一步明确。CXCR3趋化因子系统的特征和作用使其成为免疫治疗的潜在靶点,为肿瘤治疗带来新的希望。
猜你喜欢
中国生殖健康(2020年7期)2020-12-10 07:48:51
材料科学与工程学报(2016年4期)2017-01-15 13:35:48
海南医学(2016年8期)2016-06-08 05:43:00
合成化学(2015年4期)2016-01-17 09:01:11
中国医药生物技术(2015年4期)2015-12-26 08:26:34
现代检验医学杂志(2015年6期)2015-02-06 01:44:04
无机化学学报(2014年6期)2014-02-28 17:32:06
无机化学学报(2014年5期)2014-02-28 17:31:42
河南医学研究(2014年4期)2014-02-27 14:52:17
西南军医(2014年1期)2014-02-03 03:06:31
