
TNFSF13B基因与肾透明细胞癌预后及肿瘤微环境的关系
2021-09-14 07:29郑浩然蒋爱民
现代泌尿外科杂志 2021年8期
郑浩然,蒋爱民,刘 娜,傅 潇,姚 煜
(西安交通大学第一附属医院肿瘤内科,陕西西安 710061)
在世界范围内,肾癌约占成人恶性肿瘤的2%~3%,其最常见的病理类型为肾透明细胞癌(renal clear cell carcinoma,RCCC),约占肾癌病理分型的75%[1]。RCCC预后差,早期诊断困难,20%的患者初诊时已经出现转移,晚期患者5年生存率小于10%[2]。因此,找到RCCC早期诊断及预后相关的生物标志物至关重要。以培唑帕尼、舒尼替尼等为代表的靶向治疗是晚期RCCC的传统一线治疗。近年来,免疫治疗的出现在晚期RCCC的治疗中具有里程碑式意义[3],肿瘤微环境(tumor microenvironment,TME)也成为当下的研究热点。本研究基于TCGA数据库,通过生物信息学方法找到与RCCC肿瘤微环境相关的差异表达基因TNFSF13B,进一步验证了其在RCCC中的表达、对RCCC预后的影响及其与RCCC肿瘤微环境成分的关系,为RCCC的发病机制、早期诊断、综合治疗及预后评价提供了新思路。
1 资料与方法
1.1 数据来源605例RCCC病例(正常样品72例,肿瘤样品533例)的转录组RNA-seq数据和相应的临床数据均从TCGA数据库(https:∥portal.gdc.cancer.gov/)下载。
1.2 ESTIMATE评分、免疫评分和基质评分的计算
使用R语言estimate包中的ESTIMATE算法估计肿瘤微环境中免疫成分和基质成分的比率,以3种得分的形式展现:免疫评分(immune score)、基质评分(stromal score)和ESTIMATE评分。其分别与免疫成分、基质成分和两者之和呈正相关。得分越高,TME中相应成分的比率越大[4]。
1.3 生存分析及临床相关性分析使用R语言survminer包进行生存分析,生存曲线通过Kaplan-Meier法绘制,采用对数秩和检验,以P<0.05差异有统计学意义。使用Wilcoxon秩和检验或Kruskal-Wallis秩和检验进行临床相关性分析,P<0.05为差异有统计学意义。
1.4 在免疫评分和基质评分的高分组和低分组之间生成差异表达基因根据免疫评分和基质评分的中位数将肿瘤样本分为高分组和低分组。通过R语言limma包对高分组之间和低分组之间分别进行分析,筛选出差异表达基因(differently expressed genes,DEGs)[5]。以|log2FC|>1和错误发现率(FDR)<0.05作为DEGs的截断标准。
1.5 GO和KEGG富集分析通过R语言clusterProfiler、enrichplot、ggplot2软件包对筛选出的93个DEGs进行基因本体(gene ontology,GO)分析及京都基因和基因组百科全书(kyoto encyclopedia of genes and genomes,KEGG)分析,以P值和q值均小于0.05为显著性富集。
1.6 识别核心基因首先,采用STRING(https:∥string-db.org/)数据库对DEGs进行蛋白质-蛋白质相互作用网络(protein-protein interaction,PPI)构建[6]。随后,运用Cytoscape 3.8.0(http:∥www.cytoscape.org/)[7]中的cytohubba插件对PPI网络进行重构,采用交互关系置信度大于0.95的节点构建网络。其次,通过R语言survival包进行单因素Cox回归分析,分析结果在森林图中显示。最后,对PPI网络及单因素Cox回归分析中发现的DEGs绘制韦恩图以获得核心基因。
1.7 GSEA富集分析从Molecular Signatures(https:∥www.gsea-msigdb.org/gsea/msigdb/index.jsp)数据库下载Hallmark和C7(免疫相关通路)数据集作为目标集,进行GSEA富集分析[8]。用所有肿瘤标本的全部转录组数据进行GSEA富集分析,只有当NOMP<0.05且FDRq<0.06时即为显著性富集。
1.8 肿瘤浸润性免疫细胞谱及其相关性分析通过R语言CIBERSORT算法估算所有肿瘤样本中肿瘤浸润性免疫细胞的丰度分布。随后进行免疫细胞间相关性分析、免疫细胞与基因表达水平差异性和相关性分析。
1.9 HPA数据库运用HPA数据库(https:∥www.proteinatlas.org/)初步验证基因在RCCC和正常组织中的蛋白质表达情况。
2 结 果
2.1 高免疫评分与RCCC患者不良预后及淋巴结转移相关除了肿瘤细胞外,TME主要由免疫细胞和基质细胞等成分组成,在肿瘤的发生、发展过程中产生重要作用[9]。基质细胞已被证实在肿瘤的血管生成和基质重塑方面发挥重要作用[10]。而TME中不同种类的免疫细胞在抗肿瘤和促肿瘤方面发挥不同作用。越来越多的研究表明,TME中肿瘤浸润性免疫细胞水平可能作为疗效预测的指标[11]。本研究从TCGA数据库下载RCCC患者的基因表达谱数据,通过ESTIMATE算法计算免疫评分、基质评分和ESTIMATE评分,进而估计免疫细胞和基质细胞在RCCC微环境中所占的比例。对三种评分分别进行Kaplan-Meier生存分析以建立TME中免疫和基质比例与患者生存的相关性(图1)。结果表明,免疫评分高与RCCC患者预后不良相关(P=0.033,图1B)。然而,ESTIMATE评分和基质评分与患者预后无相关性(图1A和图1C)。随后,我们研究了3种评分与RCCC主要临床病理特征的关系(图2),结果显示,免疫评分高与RCCC淋巴结转移显著相关(P=0.021,图2G)。
2.2 基于免疫评分和基质评分筛选DEGs并进行GO和KEGG富集分析将RCCC样本根据免疫评分和基质评分的中位数分为高分组和低分组,构建基因表达谱热图(图3A、C),进而筛选出DEGs。结果表明,两组之间有44个上调的DEGs和49个下调的DEGs(图3B、D)。随后,我们对这些DEGs进行了GO和KEGG富集分析,以研究这些DEGs最常见的生物学过程和途径。GO富集分析结果表明,这些DEGs主要参与B细胞增殖、体液免疫应答、白细胞增殖、淋巴细胞增殖和单核细胞增殖(图3E)。KEGG富集分析结果表明,这些DEGs主要在细胞因子-细胞因子-受体相互作用、造血细胞系、原发性免疫缺陷、酮体的合成和降解等通路富集(图3F)。
2.3 通过PPI网络和单因素Cox回归分析确定核心基因我们对DEGs进行PPI网络构建和单因素Cox回归分析,以确定RCCC中潜在的与预后相关的核心基因。图4A显示了前52个基因之间的蛋白质相互作用,图4B用条形图表示按节点数排列的前30个基因。单因素Cox回归分析结果表明,23个DEGs与RCCC的预后相关(图4C)。最后,我们选择PPI网络中的前20个基因与单因素Cox回归分析筛选出的23个基因并绘制韦恩图,从而确定核心基因。结果表明,只有IGLL5和TNFSF13B重叠(图4D)。
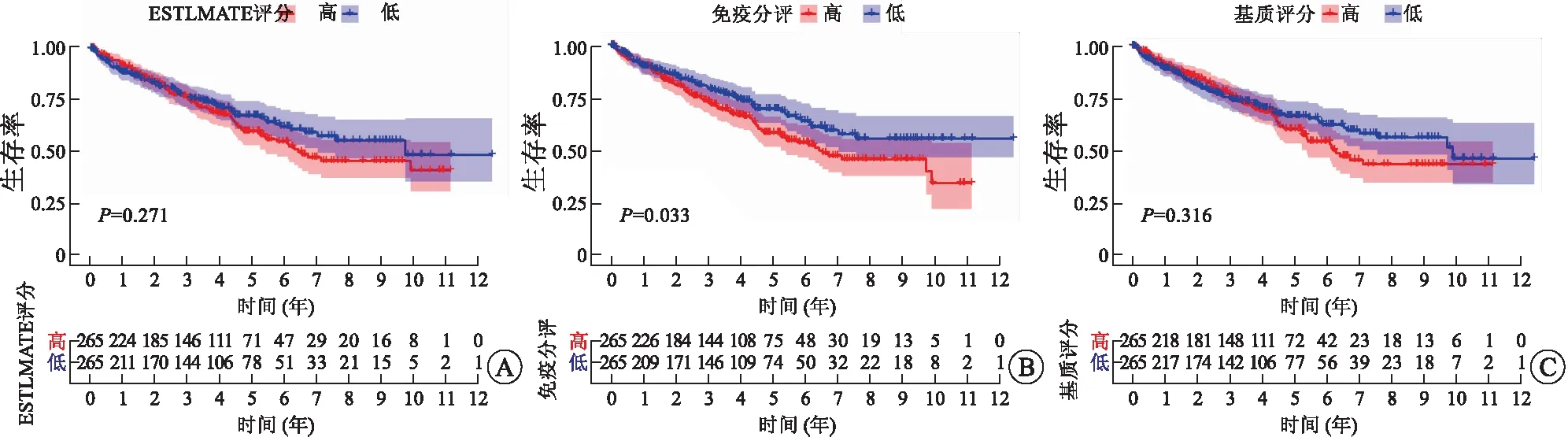
A:ESTIMATE评分;B:免疫评分;C:基质评分。
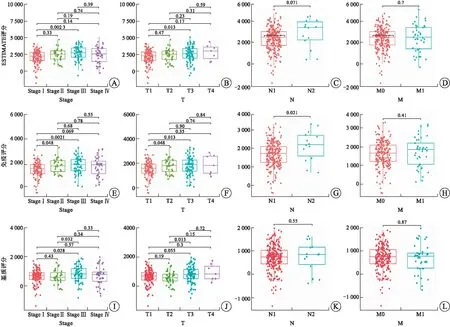
A~D:ESTIMATE评分;E~H:免疫评分;I~L:基质评分。
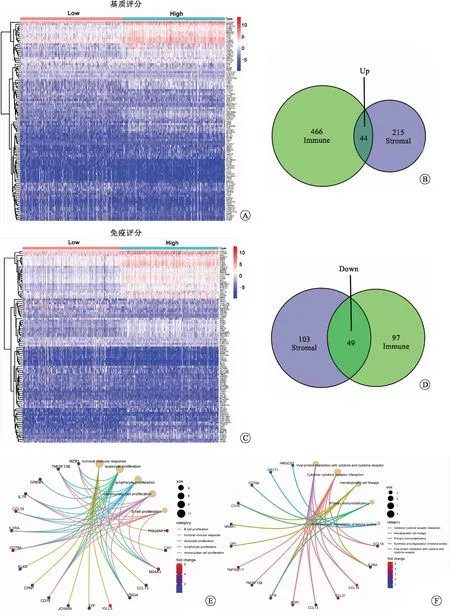
A:基质评分DEGs热图;B:免疫评分DEGs热图;C:免疫评分和基质评分共有上调DEGs的韦恩图;D:免疫评分和基质评分共有下调DEGs的韦恩图;E:DEGs的GO富集分析;F:DEGs的KEGG富集分析。
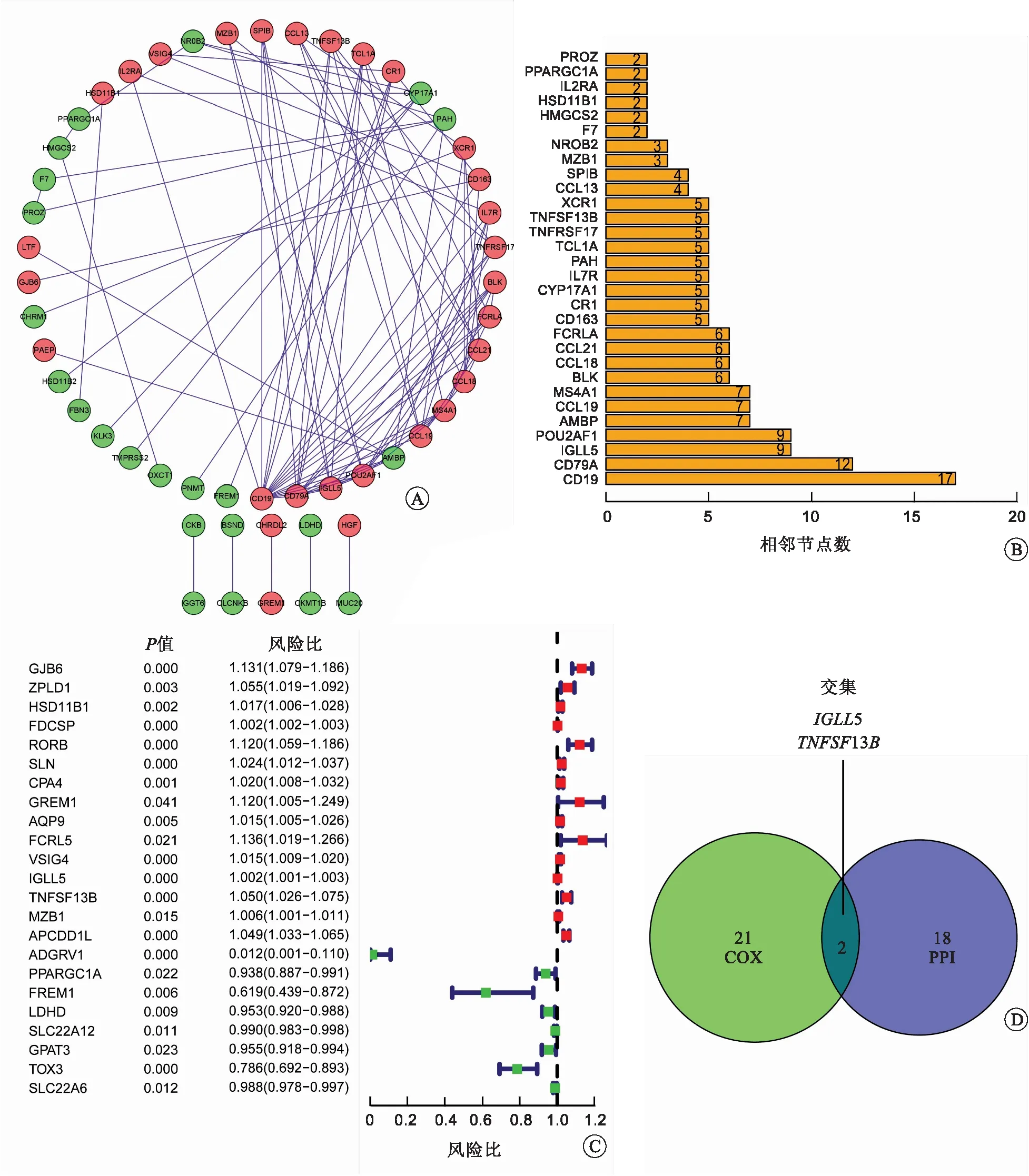
A:PPI网络;B:按节点数排列的前30个基因;C:DEGs的单因素Cox回归分析;D:PPI网络中前20个基因和单因素Cox回归分析共有基因的韦恩图。
2.4TNFSF13B的表达与RCCC预后、N分期及TME相关TNFSF13B又称B细胞激活因子(BAFF),在B细胞生存、增殖、向浆细胞分化等方面发挥关键作用[12]。作为免疫相关基因,我们进一步研究了TNSF13B表达与RCCC预后、临床病理特征和TME的关系。结果表明TNFSF13B在RCCC肿瘤组织中的表达显著升高(图5A、B)。通过HPA数据库初步验证TNFSF13B蛋白在RCCC组织中的表达高于正常组织(图8D)。生存分析结果表明TNFSF13B高表达与RCCC患者更短的总体生存率(OS)显著相关(图5C,P<0.001)。此外,我们还发现TNFSF13B的高表达与RCCC淋巴结转移相关(图5F)。随后,对C7基因集(免疫相关通路)的GSEA富集分析结果显示,TNFSF13B的高表达主要在抗原的处理和提呈、趋化因子信号通路、细胞因子-细胞因子-受体相互作用和自然杀伤细胞介导的毒性等通路富集(图6A)。此外,对Hallmark基因集的GSEA富集分析结果表明,在TNFSF13B高表达组中,多个免疫通路相关基因集,如移植物排斥反应、细胞凋亡、补体、IL2-STAT5信号通路和IL6-JAK-STAT3信号通路等显著富集(图6B)。这表明,TNFSF13B与RCCC免疫微环境存在一定的相关性。
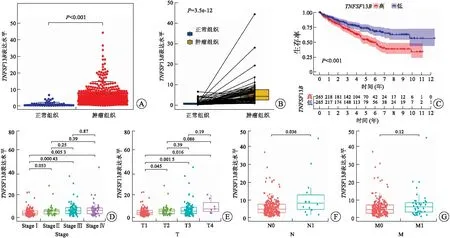
A:TNFSF13B在RCCC肿瘤组织和正常组织中的表达;B:同一患者来源的正常组织和肿瘤组织中TNFSF13B表达的配对差异性分析;C:不同TNFSF13B表达RCCC患者的生存分析;D~G:TNFSF13B表达与RCCC患者临床病理特征的相关性。
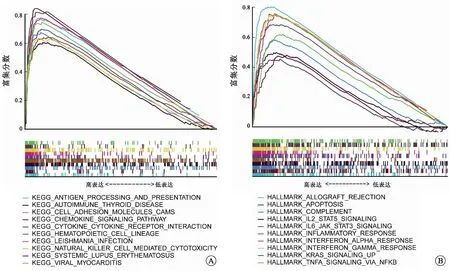
A:TNFSF13B高表达样本中C7基因集的GSEA富集分析;B:TNFSF13B高表达样本中Hallmark基因集的GSEA富集分析。
2.5TNFSF13B表达与肿瘤浸润性免疫细胞的相关性为了进一步证实TNFSF13B表达与RCCC免疫微环境的相关性,我们采用CIBERSORT算法分析了RCCC肿瘤浸润免疫细胞亚群的丰度分布(图7A),构建了RCCC样本的21种免疫细胞相关性图谱(图7B)。我们进一步绘制了免疫细胞浸润水平相对于TNFSF13B表达水平差异性的小提琴图(图8A)和免疫细胞浸润水平与TNFSF13B表达水平相关性的散点图(图8B)。差异性和相关性分析结果表明,共11种肿瘤浸润免疫细胞与TNFSF13B表达相关(图8C)。其中未成熟B细胞、静息CD4+记忆T细胞、活跃树突状细胞、静息肥大细胞与TNFSF13B表达呈负相关。CD8+T细胞、活跃的CD4+记忆T细胞、Tfh细胞、Treg细胞、γδT细胞、静息的NK细胞、嗜酸性粒细胞与TNFSF13B表达呈正相关。这些结果进一步证实TNFSF13B表达会对RCCC肿瘤免疫微环境产生一定影响。
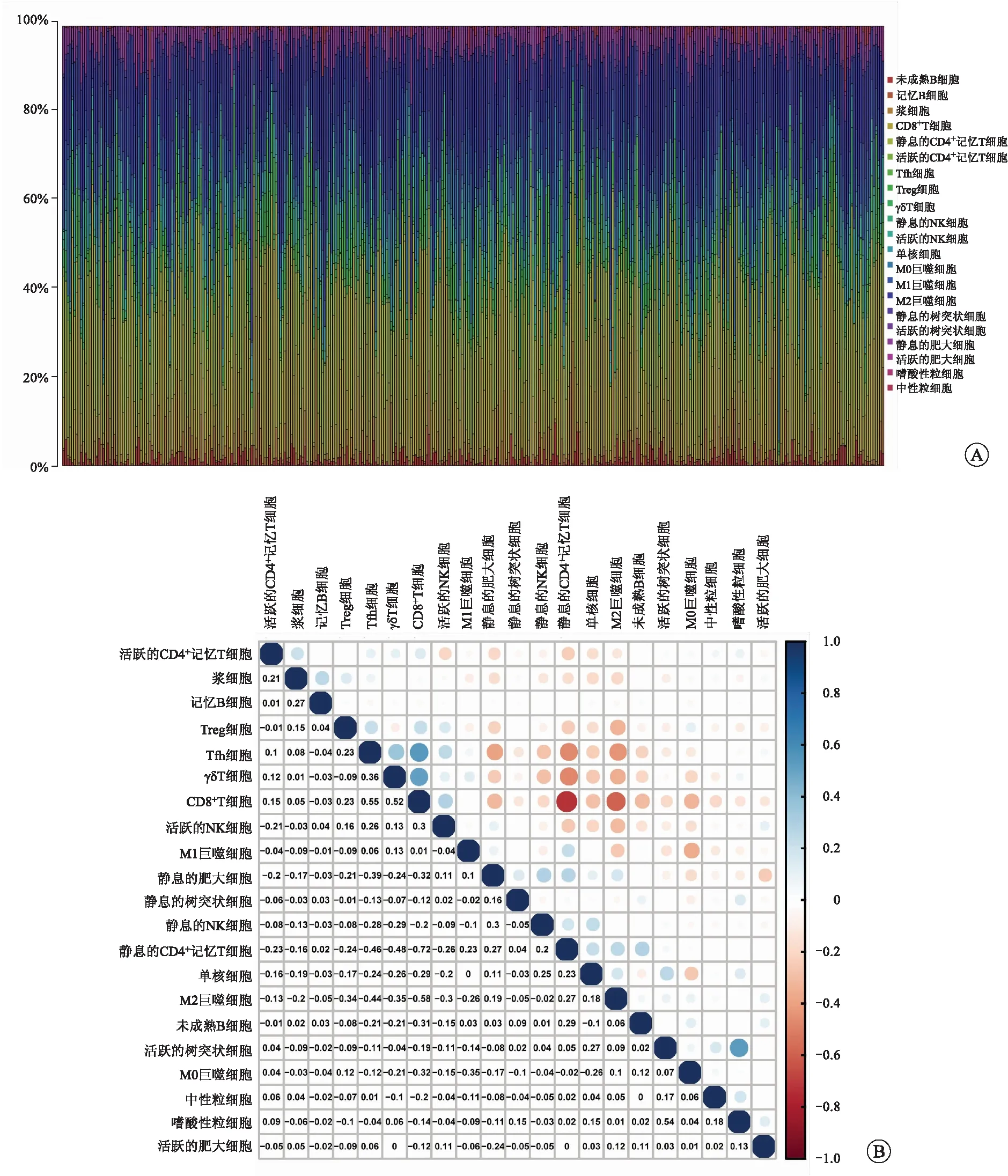
A:RCCC样本中21种肿瘤浸润性免疫细胞比例;B:RCCC样本中21种肿瘤浸润性免疫细胞的相关性。
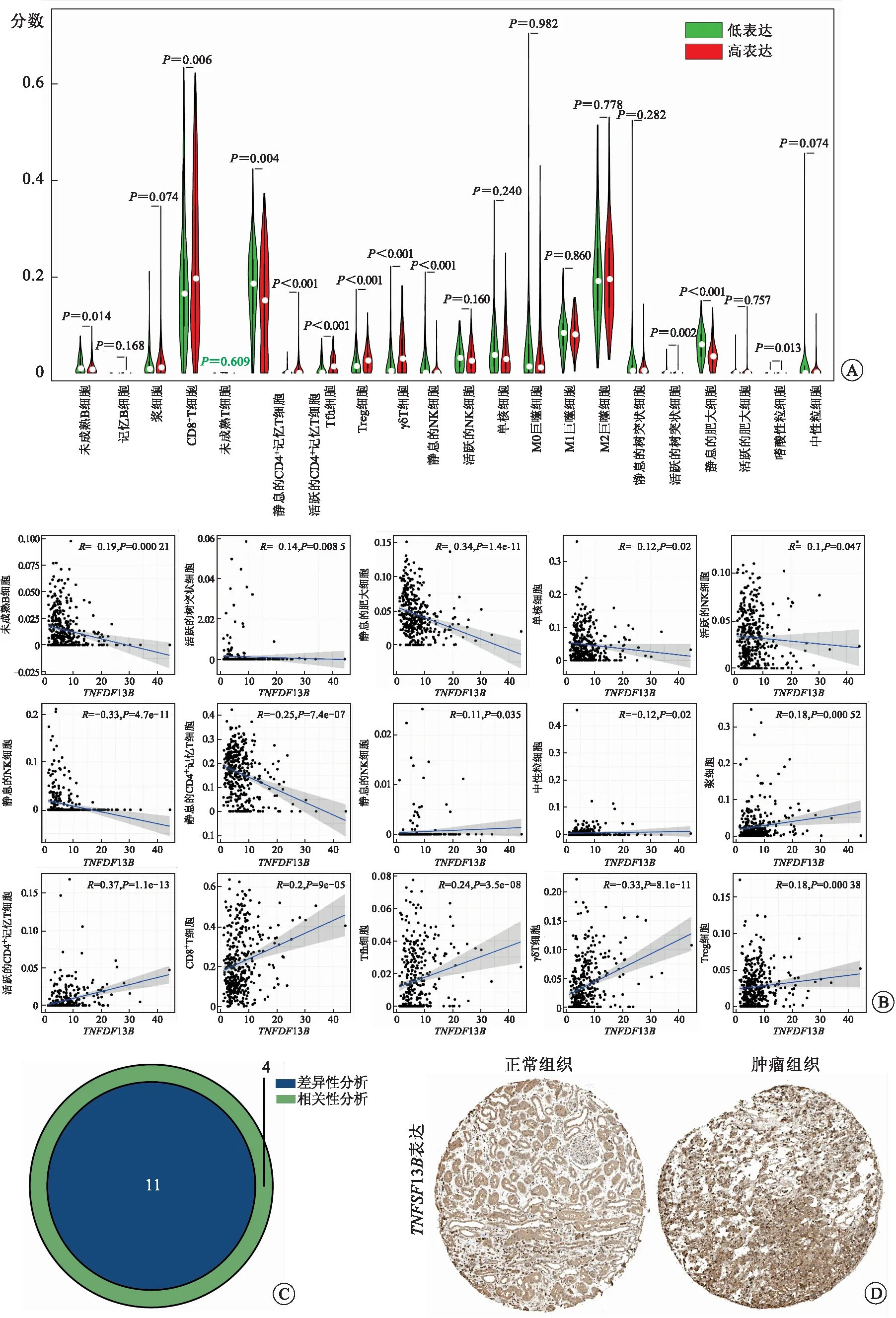
A:RCCC中21种肿瘤浸润性免疫细胞比例相对于TNFSF13B表达水平差异性分析的小提琴图;B:RCCC中15种肿瘤浸润性免疫细胞与TNFSF13B表达水平相关性的散点图;C:差异性分析和相关性分析共有的肿瘤浸润性免疫细胞韦恩图;D:TNFSF13B蛋白在RCCC肿瘤组织和正常组织中的表达。
3 讨 论
大量研究表明RCCC是免疫原性恶性肿瘤,TME中存在大量浸润性免疫细胞,如CD8+T细胞、CD4+T细胞、巨噬细胞、NK细胞、髓样细胞(myeloid-derived suppressor cells,MDSCs)等[13]。然而,CD8+T细胞和CD4+T细胞的大量浸润却与RCCC的高分级和生存期缩短相关。研究表明,CD8+T细胞的高增殖率与RCCC患者生存期延长相关,这表明免疫细胞的功能状态是决定其抗肿瘤活性的关键因素。RCCC中肿瘤浸润性淋巴细胞无法发挥抗肿瘤功能可能是与Treg细胞、MDSCs等免疫抑制性细胞的大量浸润相关[14]。RCCC的免疫抑制状态可以通过应用靶向免疫检查点抑制剂的负向调节作用打破,恢复抗肿瘤免疫反应。RCCC中30%的患者存在PD-L1的过表达[15],TME中亦经常发现PD-1阳性的巨噬细胞、MDSCs和NK细胞。巨噬细胞中PD-1的表达与M2型巨噬细胞的极化以及IL-6的产生增加相关,从而导致免疫抑制及肿瘤进展[16]。免疫检查点抑制剂在RCCC的治疗过程中显示出良好的疗效,目前,免疫治疗以及免疫治疗联合靶向治疗已经成为晚期RCCC患者的标准治疗模式。尽管如此,部分RCCC患者对免疫治疗响应差,找到能够影响RCCC肿瘤微环境及预测免疫治疗疗效的生物标志物至关重要。
本研究基于TCGA数据库发掘出的与RCCC患者预后及TME相关的关键基因TNFSF13B又称B细胞激活因子(B cell activating factor,BAFF),是肿瘤坏死因子(tumor necrosis factor,TNF)家族成员之一,在B细胞增殖分化过程中发挥重要作用。血液中可溶性BAFF的升高与系统性红斑狼疮等自身免疫性疾病的发生密切相关[17]。自发现以来,BAFF在肿瘤发生发展过程中的作用一直是研究的热点。由于BAFF在维持B细胞和浆细胞的存活方面发挥重要作用,血清中BAFF水平的升高与多发性骨髓瘤和B淋巴细胞恶性肿瘤患者不良预后相关[18-19]。目前,美国食品药品监督管理局(food and drug administration,FDA)批准的抗BAFF抗体贝利木单抗(Belimumab)只适用于系统性红斑狼疮的治疗,其在B细胞淋巴瘤的疗效仍在临床试验过程中[20]。在实体瘤中,已有研究表明,胰腺癌、肾癌及神经内分泌癌等肿瘤中TNFSF13B表达升高,且与患者不良预后相关[21-23]。RIHACEK等[24]的研究表明BAFF可能通过促进炎症反应及胰岛素抵抗导致癌症患者晚期恶病质的出现。最近,YARCHOAN等[25]的研究表明,BAFF在TME中发挥双重作用:既可以增强B细胞对CD4+Th细胞的抗原提呈能力,增强TME中Th1细胞的抗肿瘤活性,又能够导致TME中Treg细胞的增加,从而介导肿瘤免疫逃逸。我们的研究发现Treg细胞浸润水平与TNFSF13B表达呈正相关,这说明TNFSF13B高表达可能与RCCC免疫逃逸及免疫抑制状态相关。CD8+T细胞、活跃的CD4+记忆T细胞等抗肿瘤免疫细胞水平虽然与TNFSF13B表达呈正相关,但其功能可能处于抑制状态,无法发挥抗肿瘤效应。TNFSF13B对RCCC肿瘤微环境影响的具体机制仍有待于更详细、更具体的体内外研究的进一步开展。
综上所述,TNFSF13B基因高表达与RCCC患者的不良预后显著相关,并对RCCC肿瘤微环境产生一定影响。本研究基于公共数据库进行研究,存在一定的局限性,如样本量较小、未通过真实世界进行验证等。但本研究找到了与RCCC预后及TME相关的关键基因,为RCCC的发病机制、早期诊断、预后评估及治疗策略提供了一定的依据,为新型药品的研发及RCCC基础与临床试验提供了新思路。TNFSF13B有望成为RCCC新的潜在治疗靶点和预后相关的生物学标志物。
猜你喜欢
表面技术(2022年9期)2022-09-27
今日农业(2021年9期)2021-11-26
果农之友(2021年2期)2021-03-24
中华养生保健(2021年18期)2021-02-13
健康博览(2017年8期)2017-12-05
科技资讯(2017年7期)2017-05-06
农村百事通(2017年6期)2017-03-30
热带农业工程(2014年6期)2015-01-28
现代农业科技(2009年19期)2009-03-20
中学生数理化·八年级数学华师大版(2008年3期)2008-08-26
