
2020年版GCP实施后可疑且非预期严重不良反应报告分析*
2021-09-12 05:22汪华蓉
中国药业 2021年17期
王 静,杜 彪,汪华蓉
(重庆大学附属三峡医院药学部,重庆404000)
可疑且非预期严重不良反应(SUSAR)是药物临床试验中安全性信息的重要组成部分。国家相关部门近年来发布了系列临床试验法律法规,逐步优化了报告流程,明确了报告要求[1]。2020年版《药物临床试验质量管理规范》(GCP)于2020年7月1日起施行,增加了SUSAR的定义[2],明确为临床表现的性质和严重程度超出了试验药物研究者手册、已上市药品的药品说明书或产品特性摘要等已有资料信息的SUSAR。第四十八条新增:“申办者应当将可疑且非预期严重不良反应快速报告给所有参加临床试验的研究者及临床试验机构、伦理委员会”。2020年7月至10月收到1 323份SUSAR报告,数量远高于2016年至2017年接收的299份[3],SUSAR数量的骤增对药物临床试验机构办公室、药物临床试验伦理委员会、研究者提出了严峻挑战。本研究中分析了2020年7月至10月临床试验机构收到的SUSAR报告,从药物临床试验机构层面分析和掌握现状,为高质、高效处理SUSAR报告和降低受试者风险提供参考。现报道如下。
1 资料与方法
1.1 资料来源
资料来源于2020年7月至10月由申办者报告至药物临床试验机构办公室的1 323份SUSAR报告,其中涉及本院受试者3份,非本院受试者1 320份;涵盖的专业与科室包括肿瘤专业1 080份,内分泌专业233份,肝炎专业4份,神经内科5份,消化内科1份。
1.2 方法
回顾性分析1 323份SUSAR个例报告,从试验项目分期、试验用药物的注册分类、事件转归情况、事件性质(类别)等因素进行统计与分析。将SUSAR事件的结果/转归分为6类状态:痊愈、好转/缓解、未好转/未缓解/持续、痊愈伴后遗症、致死、未知[4]。
2 结果
2.1 在试验项目分期中的分布
1 323份SUSAR报告中,发生于Ⅱ期药物临床试验项目245份(18.52%),Ⅲ期药物临床试验项目1 078份(81.48%)。Ⅲ期临床试验中的SUSAR报告份数远高于Ⅱ期临床试验。
2.2 试验用药物的注册分类及转归情况分布
1 323份SUSAR个例报告中,试验用药物的注册分类共6大类别,化学药品1类54份(4.08%),化学药品5类9份(0.68%),生物制品1类1 212份(91.61%),生物制品2类43份(3.25%),生 物 制 品7类2份(0.15%),生物制品9类3份(0.23%)。可见,生物制品占比远高于化学药品,且生物制品1类与化学药品1类显著高于同类别其他药品。详见表1。
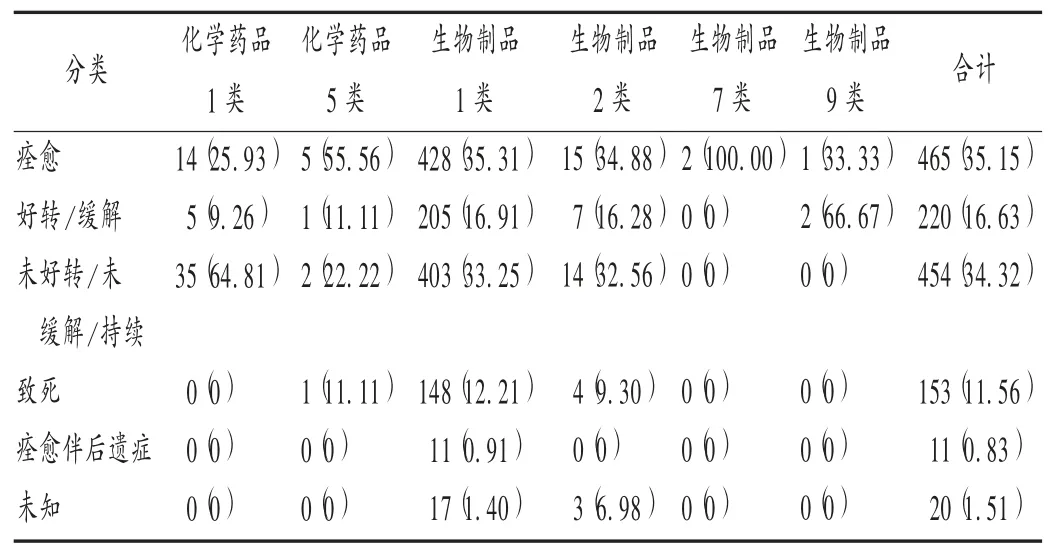
表1 SUSAR试验用药物的注册分类与转归情况分布[份(%)]Tab.1 Distribution of registration classification and outcomes of trial drugs in SUSAR reports[n(%)]
分析6种转归情况,结局为未好转/未缓解/持续与致死的SUSAR试验用药物中,化学药品1类共35份(64.81%),生物制品1类共551份(45.46%),生物制品2类18份(41.86%);生物制品2类总份数虽远低于1类,但未好转/未缓解/持续率与致死率差异不明显;结局为致死的SUSAR试验用药物中,生物制品1类148份(12.21%),生物制品2类4份(9.30%),化学药品5类1份(11.11%)。详见表1。转归为致死事件中的主要原因有肺炎、肺部感染、医院获得性肺炎、消化道出血、心肌梗死、发热、多器官衰竭。
2.3 性质(类别)分布
SUSAR的性质分为致死或危及生命、非致死或危及生命[5]。分析结果显示,致死或危及生命的SUSAR 272份(20.56%),非致死或危及生命的SUSAR 1 051份(79.44%)。272份致死或危及生命的SUSAR报告中,事件名称描述率较高的依次为不明原因死亡、心肌梗死、上消化道出血、心脏停搏、脂肪酶增高。详见表2。

表2 SUSAR的性质(类别)分布(份)Tab.2 Distribution of nature(category)of trial drugs in SUSAR reports(n)
3 讨论
Ⅲ期临床试验中的SUSAR报告份数高于Ⅱ期临床试验,分析原因可能为:1)临床试验机构总体Ⅲ期在研项目数量多于Ⅱ期,基数低;2)当临床试验进行到Ⅲ期时,申办者常会探索试验用药物在多个适应证中的安全性和有效性,往往对同一药物会同时发起多项临床试验,故在同一时间段、同一个试验用药物的试验项目数量会高于Ⅱ期;3)Ⅱ期临床试验整体设计入组的受试者样本量较低。以上因素均使Ⅱ期临床试验阶段暴露人群有限,故发现和获知的SUSAR较少。药品不良反应(ADR)是客观存在的,从Ⅲ期临床试验中SUSAR份数也能体现,故不能因为Ⅱ期临床试验的SUSAR数量少就降低关注度,反而在早期阶段应更加予以重视。
全部在研药物临床试验项目中,生物制品类与化学药品类项目数量比例为4∶3,结合药品注册分类分析结果,生物制品的SUSAR报告份数显著高于化学药品,分析原因可能为,该院大部分生物制品为抗肿瘤药物,受试者常有复杂的合并疾病,预期生存时间较其他疾病短,故出现SUSAR的概率高;另一方面,生物制品的剂型、给药途径、成分复杂等自身特点决定了其可能导致的过敏反应、输液反应等ADR高于化学药品。
由于1类创新药为国内外均未上市的药品,临床前研究资料、临床试验数据有限,试验暴露人群极少,故生物制品1类与化学药品1类的SUSAR报告份数和发生率均显著高于同类的其他注册类别。对于生物制品1类,其SUSAR报告总份数、致死率、未好转/未缓解/持续率与致死率均处于首位,故生物制品1类应作为重点风险监控品种,且在试验前的方案设计、风险控制措施制订时应特别重视,需建立切实可行的早期风险预警机制。虽然化学药品1类的数量低于生物制品1类,但其痊愈率与好转/缓解率均低于生物制品1类,故为降低和避免受试者面临的风险,应同样将化学药品1类纳入重点监控品种。对于1类新药的临床试验,建议将SUSAR报告中的转归为未好转/未缓解/持续、致死信息,以及与试验用药物相关性级别作为风险信号,当出现此类信号时,研究者应予以重点审核,并评估对受试者的影响;同时建立追踪机制,一方面追踪SUSAR的进一步转归,另一方面重点追踪对受试者的再次知情及相关受试者保护措施的执行情况。
加入人用药品注册技术要求国际协调会议(ICH)后,将吸引更多跨国企业和国外中小型企业到中国开展临床试验,其中不乏高风险的更具有挑战性的试验项目[6]。同时,由于我国药物临床试验机构备案制的实施,新的药物临床试验机构开展药物临床试验的经验较少,对GCP意识较薄弱[7]。因此,结合以上分析及本院经验,为提高SUSAR审核的时效性和有效性,可在临床试验准备阶段制订良好的项目安全性监测计划[8],药物临床试验机构有必要建立高风险品种风险预警机制[3],制订重点风险监控品种目录,明确风险信号识别措施[9]。生物制品1类、化学药品1类可列入重点风险监控品种目录,在准备阶段就应重点审核其风控计划与措施是否可行和合理。研究者手册是临床试验期间风险管理的重要工具,研究人员在启动前需重点审阅[10]。在试验中,可定期对收到的SUSAR进行汇总分析,将致死或危及生命发生率、痊愈与好转率作为风险识别信号,定期更新重点风险监控品种目录,把致死或危及生命发生率高的药物、痊愈与好转率低的品种纳入重点风险监控品种目录。伦理委员会担负着保护受试者的重要职责,可结合汇总分析结果评估临床研究的受益风险比,确保让受试者获得对等的信息,充分了解研究中某些可能的潜在伤害[11]。
提高试验期间SUSAR快速报告的质量和完善药物警戒体系还需着重考虑人员和方法规章因素,临床试验机构应确认相关措施是否被执行,以及受试者权益是否被妥善保护[12-13]。在整理SUSAR报告中发现,目前仍有部分申办者未按规范要求进行SUSAR快速报告,存在报告不及时、漏报情况,故需进一步强调申办者的主体责任,提高药物临床试验期间SUSAR快速报告的质量[14],同时药物临床试验机构应培养专职、专业的工作人员,以促进对试验安全信息的管理[15]。SUSAR是评价药物安全性的重要数据来源,也是起草和撰写药品说明书安全性信息的重要依据[16],第一时间及时掌握安全性信息,可避免因对已出现的风险识别过晚,让受试者暴露于不必要的风险之中,故药物临床试验机构有必要建立一定的惩罚机制,督促申办者及时、规范报告,保障受试者安全。
猜你喜欢
猪业科学(2022年10期)2022-11-03
吉林畜牧兽医(2022年7期)2022-07-20
猪业科学(2022年2期)2022-04-21
猪业科学(2022年1期)2022-03-24
教学月刊(小学版)(2020年29期)2020-12-30
中国外汇(2020年10期)2020-11-25
科学与财富(2017年35期)2018-01-29
消费导刊(2018年24期)2018-01-28
读写算·高年级(2017年4期)2017-04-15
中西医结合心血管病电子杂志(2014年1期)2014-08-11
