
铁死亡相关机制及其在肾脏相关疾病中的研究进展
2021-09-09 01:07孙佳宾岳明豪夏舜尧修有成
激光生物学报 2021年4期
孙佳宾,岳明豪,刘 赞,夏舜尧,修有成
(哈尔滨医科大学附属第一医院泌尿外科,哈尔滨 150001)
细胞死亡对机体正常生长发育、维持动态平衡和预防癌症等增殖性疾病至关重要。研究人员曾经认为细胞凋亡是唯一的细胞死亡形式,但随着研究的深入,他们又发现了不同于细胞凋亡的死亡形式,如凋亡、坏死性凋亡、细胞自噬、细胞焦亡以及铁死亡(ferroptosis)[1]。铁死亡是一种全新的细胞死亡形式,与之前发现的细胞死亡方式完全不同(表1),其主要特点为铁离子累积与脂质过氧化的发生。近年来,越来越多的研究表明铁死亡与多种疾病的发生发展有关,如帕金森病、缺血再灌注损伤等[2],此外,铁死亡与肾脏相关疾病有密切关系,但铁死亡与肾脏相关疾病的确切机制尚未探明。本文将对铁死亡相关机制及其在肾脏相关疾病中的研究进展进行综述,并对其发展前景进行展望。

表1 五种细胞死亡形式的比较Tab. 1 Comparison of five types of cell death
1 铁死亡的发现过程及其特点
在2003年的一项研究中,Dolma等[3]发现了Erastin和RAS选择性致死蛋白3(RAS-selective lethality protein 3,RSL3)对RAS突变细胞具有选择性致死作用。2007年,Yagoda等[4]发现由Erastin所导致的细胞死亡是与细胞凋亡、坏死和自噬不同的细胞死亡形式,其形态学特征主要为线粒体完整性的消失。2008年,Chen等[5]提出了该死亡形式与细胞内活性氧(reactive oxygen species,ROS)水平升高有关,并发现该过程可通过抑制细胞摄取铁来终止。2012年,Dixon等[6]正式命名了该种细胞死亡形式——铁死亡。铁死亡在形态学以及生化特征上与细胞凋亡等细胞死亡形式不同。在形态学上,铁死亡主要表现为细胞膜断裂和出泡、线粒体嵴缩小、线粒体双层膜密度增加。在生化特征方面,铁死亡主要表现为ROS的聚集、脂质过氧化物含量的增多以及细胞内铁离子的累积[6]。
2 铁死亡的相关机制
随着研究的深入,人们发现铁离子代谢紊乱、胱氨酸/谷氨酸反向转运体(cystine/glutamate antiporter,system xc-)的抑制、脂质过氧化物的累积、谷胱甘肽过氧化物酶4(glutathione peroxidase 4,GPX4)的活性下降为铁死亡的主要机制(图1)。
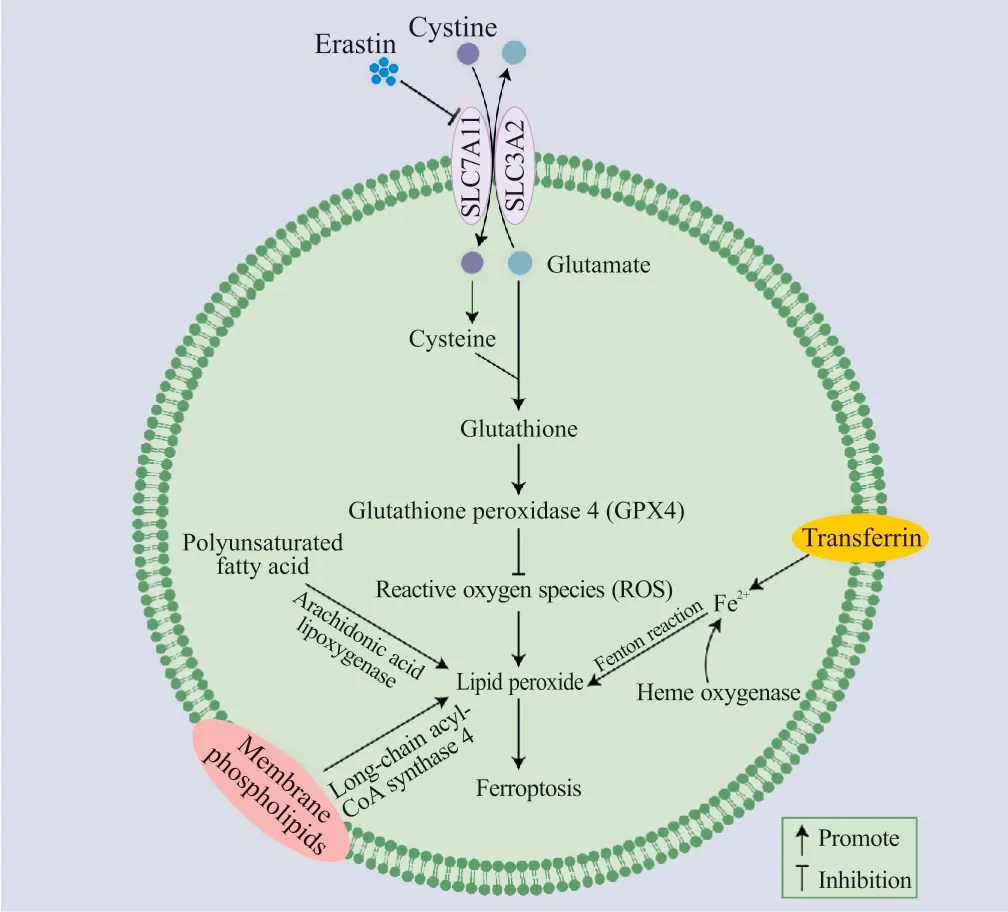
图1 铁死亡发生的主要机制Fig. 1 Main mechanism of ferroptosis
2.1 铁代谢异常
铁在人体中是一种参与多项生理活动发生的重要元素,如同蛋白质形成血红蛋白、参与氧的运输、构成人体所必须的酶。铁离子的累积是铁死亡的主要特征之一。但是,目前铁在铁死亡发生过程中的具体机制尚未被完全揭示[5]。先前的研究证明,铁螯合剂去铁胺可以通过减少细胞内铁离子含量进而抑制铁死亡的发生[6],而细胞内铁离子含量的增多可促进铁死亡的发生[7],表明铁离子累积在铁死亡发生过程中起重要作用。Alvarez等[8]的研究发现,铁硫簇生物合成酶(iron-sulfur cluster biosynthetic enzyme,NFS1)可以通过抑制细胞内铁水平的升高进而抵抗铁死亡的发生。铁硫簇是细胞铁水平的传感器,在高氧条件下,铁硫簇会快速降解,当细胞内铁硫簇水平过低时,细胞内储存铁的分子会释放更多的铁,从而发生铁死亡。而NFS1可以从半胱氨酸中吸收更多的硫元素,使铁硫簇总量增加,进而抑制铁离子的释放,抑制铁死亡的发生。Fang等[9]和Chang等[10]的研究中均观察到,血红素加氧酶1(heme oxygenase 1,HMOX1)可通过促进血红素分解释放铁离子,从而诱发铁死亡的发生。综上可知,虽然铁离子在铁死亡中的作用机制仍不完全清楚,但铁代谢紊乱在铁死亡发生过程中的关键作用是毋庸置疑的。
2.2 GPX4
GPX4是已知的唯一能够直接清除脂质过氧化物的酶,可以谷胱甘肽为辅助因子将有害的脂质过氧化物还原为无害的物质,从而打断脂质过氧化反应的发生,进而抑制铁死亡[11]。其具体化学机制为GPX4在脂质过氧化物中发生亲核取代,将脂质过氧化物还原为脂质醇,从而抑制脂质过氧化的发生[12]。Yang等[13]证明了GPX4是铁死亡的关键调节因子,并且可以在小鼠肿瘤移植瘤中通过抑制GPX4诱导铁死亡。Gaschler等[14]的研究发现,五元环过氧化物1,2可以通过间接失活GPX4来增加细胞对铁死亡的敏感性。由此可见,GPX4是铁死亡的调控过程中的重要因子。
2.3 脂质过氧化
脂质过氧化在铁死亡发生过程中起驱动作用,可以通过非酶促的方式完成,也可通过酶促反应发生。例如,铁可以通过增加花生四烯酸脂加氧酶(arachidonate lipoxygenase,ALOX)的活性来引起铁死亡。ALOX是一类非血红素的含铁酶,可以催化多不饱和脂肪酸(polyunsaturated fatty acids,PUFAs)的氧化生成丙二醛和4-羟基壬烯醛等产物,进而促进细胞铁死亡的发生[15]。Imai等[11]在研究中使用经过降低PUFAs含量处理的细胞诱导铁死亡,发现经过处理后的细胞脂质过氧化水平降低会导致其铁死亡敏感性大幅度降低。Yan等[16]发现,位于内质网上的氧化还原酶原叶绿素酸酯氧化还原酶和细胞色素B5还原酶1将来源于还原型辅酶2[NAD(P)H]的电子传给氧气生成过氧化氢,并同二价铁离子发生了芬顿反应,形成了活跃的羟基自由基,造成了脂质过氧化,导致了细胞膜的破坏,从而发生铁死亡,揭示了铁死亡发生过程中膜损伤的机制。Doll等[17]发现长链酯酰辅酶A合成酶4(long-chain acyl-CoA synthetase 4,ACSL4)是GPX4失活时发生铁死亡的关键物质。在GPX4失活的情况下,ACSL4仍然可以促进脂质过氧化,从而促进细胞铁死亡的发生。因此,脂质过氧化是驱动铁死亡的关键因素。
2.4 system xc-
system xc-是一种细胞表面钠离子非依赖性的胱氨酸-谷氨酸逆向转运蛋白,由12次跨膜转运蛋白溶质载体家族7成员11(solute carrier family 7 member 11,SLC7A11)通过二硫键连接到单次跨膜调节亚基溶质载体家族3成员2(solute carrier family 3 member 2,SLC3A2)上组成,可介导胱氨酸和谷氨酸以1∶1的比例在细胞内外交换。胱氨酸在细胞内被还原为半胱氨酸之后合成谷胱甘肽,谷胱甘肽在谷胱甘肽过氧化物酶的作用下还原活性氧和活性氮。Erastin可通过抑制system xc-介导的胱氨酸摄取损害细胞的抗氧化防御,从而促进ROS的累积,进而促进铁死亡的发生[3]。Chang等[10]通过研究发现,SLC7A11的增加能够明显抑制铁死亡的发生。在另一项研究中,Liu等[18]则发现抑制SLC7A11的表达能够使细胞对铁死亡更加敏感。随着研究的深入,研究人员发现转录激活因子3[19]、beclin1[20]、卵巢肿瘤相关蛋白酶B1[21]等因子同样可通过抑制system xc-促进ROS的累积,进而促进铁死亡的发生。由此可见,system xc-在铁死亡的发生过程中起重要作用。
2.5 p53
p53是一种重要的抑癌基因,其所介导的细胞周期停滞和细胞凋亡在阻碍癌症发生的过程中起重要作用。虽然p53的一些靶点已被确定,但目前p53抑制肿瘤的机制还不完全清楚[18]。Jiang等[22]的研究发现,p53可通过抑制SLC7A11的表达来抑制胱氨酸的摄取,使谷胱甘肽生成量大量减少,从而促进细胞铁死亡。Chu等[23]研究发现,敲除花生四烯酸12后铁死亡的发生被特异性阻断,因此可以证明,p53通过抑制SLC7A11间接激活了ALOX12的功能,从而导致ROS应激后花生四烯酸12依赖性的铁死亡。其他研究表明,p53能抑制铁死亡的发生,如使用nutlin3恢复野生型p53活性可以使纤维肉瘤、肾癌和骨肉瘤免于抑制System xc-所致的铁死亡[24]。Xie等[25]的研究发现,p53可通过阻断二肽基肽酶-4的活性抑制Erastin诱导的肿瘤细胞的铁死亡敏感性。综上可知,p53在铁死亡中起重要作用,其机制较为复杂,亟待深入研究。
2.6 其他机制
电压依赖性阴离子通道(voltage-dependent anion channels,VDACs)是位于线粒体外膜的一种通道蛋白,在线粒体和其他细胞器通信过程中起重要作用[26]。Yagoda等[4]发现,Erastin可通过直接与VDAC2/3结合改变线粒体膜外膜的通透性,从而降低NADH的氧化速率,进而诱导细胞铁死亡。
铁死亡抑制蛋白1(ferroptosis suppressor protein1,FSP1)在既往的研究中被认为具有促进细胞凋亡的作用。Doll等[27]研究发现,FSP1豆蔻酰化后可通过NAD(P)H还原辅酶Q10抑制脂质过氧化,从而抑制铁死亡的发生。此外pi3k-akt-mtor[28]、p62-Keap1-NRF2 [17]信号通路也在铁死亡发生过程中起到负性的调控作用。
综上可知,铁死亡的相关研究尚处于初步阶段,铁死亡的具体机制尚未完全清楚,后续研究可以着手于探究铁死亡的具体机制,进而找到更多调控铁死亡发生的方式,以期应用于临床。
3 铁死亡在肾脏相关疾病中的研究
先前的研究表明,铁死亡在多种疾病中起重要作用[29]。同样,在肾脏相关疾病中也有铁死亡的发生。本文对铁死亡在不同肾脏疾病进展中作用的最新研究进行总结,为这些疾病的治疗和预防提供更多信息。
3.1 肾癌
肾细胞癌简称肾癌,是肾最常见的恶性肿瘤,占肾恶性肿瘤的85%~90%,根据病理分型可分为透明细胞癌、嫌色细胞癌、肾乳头状细胞癌、髓样癌以及未分化癌[30]。据流行病学相关数据统计,肾细胞癌的发病率仅次于膀胱肿瘤,在我国泌尿生殖系统肿瘤中排第二位,且呈逐年上升趋势[31]。肾细胞癌的主要治疗方法为手术治疗以及放化疗。虽然近来在肾细胞癌的治疗方法上取得了一定的进步,但预后仍不甚理想,因此迫切需要探索其治疗的新靶点。大量研究表明,诱导细胞铁死亡可能是肾细胞癌治疗的新方向。Yang等[13]在研究中检测了117种癌细胞对Erastin诱导的铁死亡的敏感性,发现弥漫性大B细胞淋巴瘤和肾透明细胞癌(clear cell renal cell cancer,ccRCC)对GPX4调节的铁死亡特别敏感。Zou等[32]研究发现,GPX4抑制剂对透明细胞癌杀伤性较强,GPX4的减少是铁死亡发生的关键因素。他们进一步研究发现,肾癌细胞中高表达的缺氧诱导因子(hypoxia-inducible factor,HIF)通路中的低氧诱导因子2α通过hilpda富集不饱和脂肪酸进而增强ccRCC的铁死亡敏感性,希佩尔林道(von hippel-lindau,VHL)基因是ccRCC中主要的抑癌因子。Miess等[33]发现,外源性高表达细胞内VHL基因能够减少细胞内脂质氧化物,从而抑制铁死亡的发生。该研究证实了VHL是调控ccRCC铁死亡敏感性的重要基因,证明了VHL诱导的铁死亡是治疗ccRCC的潜在靶点。Mou等[34]通过生物信息学手段发现,ccRCC中核受体辅激活蛋白4(nuclear receptor coactivator 4,NCOA4)的表达降低,并与ccRCC患者不良预后相关,而NCOA4与铁转运密切相关,其减少将导致肿瘤细胞对铁死亡的敏感性降低。因此,通过靶向NCOA4促进铁死亡可能是治疗ccRCC的有效方法。然而该结论仅仅通过生物信息分析论证,并不完全可靠,需要进一步的试验论证其有效性。青蒿琥酯(artesunate,ART)是一种来源于中药青蒿的化学物质,在多种肿瘤中显示出抗肿瘤作用[35]。Markowitsch等[36]的研究发现,ART可通过诱导耐药肾癌细胞铁死亡,显著抑制肾癌细胞的进展。这表明ART有希望作为一种有效的用于耐药肾细胞癌患者治疗的新药物,以解决肾细胞癌耐药的问题。
遗传性平滑肌瘤病肾癌是一种延胡索酸水化酶(fumarate hydratase,FH)胚系突变来源的常染色体显性遗传病。其发病率低,多为单发,但多半在首诊时即出现转移,中位生存率为24个月[37]。虽然在其发病机制等方面取得了一定进展,但其治疗手段仍以手术治疗为主,且治疗效果较差,因此寻找更佳的治疗手段对延长病人生存期非常重要。Michael等[38]的研究发现,遗传性平滑肌瘤病肾癌细胞由于FH的失活,其中的GPX4会琥珀酸化,进而其活性降低,同时细胞内核因子E2相关因子2(nuclear factor E2 related factor,NRF2)活性增加,从而避免铁死亡的发生。研究人员提出,未来的研究可以探索可应用于人体的NRF2活性抑制剂,或者研发出针对琥珀酸化的GPX4的抑制剂,促使遗传性平滑肌瘤病肾癌细胞的铁死亡,诱导肾癌细胞铁死亡,为治疗该疾病提供更好的选择。这可能是精准治疗肾癌的新方向。
3.2 多囊肾
常染色体显性遗传性多囊肾病(autosomal dominant polycystic kidney disease,ADPKD)是最常见的遗传性肾病。多囊肾的典型表现是患者双侧肾出现大量肾囊肿,且囊肿随着年龄增长不断扩大,压迫正常的肾脏组织,导致50%以上的患者发展为终末期肾病[39]。目前多囊肾的治疗手段为对症治疗,并不能延缓病情进展,因此,找到针对多囊肾发病机制的治疗手段尤为重要。Schreiber等[40]发现多囊肾上皮分泌氯化物,氯化物通道的囊性纤维化跨膜电导调节剂(cystic fibrosis transmembrane conductance regulator,CFTR)和跨膜蛋白16A(transmembrane protein 16A,TMEM16A)促进脂质氧化,从而促进多囊肿的增大,而GPX4等活性氧清除剂可使多囊肾增长速度减慢,铁抑素-1同样也能显著地延缓囊肿的扩大。该试验证实了在多囊肾发展过程中铁死亡起促进作用。
3.3 急性肾损伤
急性肾损伤(acute kidney injury,AKI)是一种可由多种原因导致的以急性肾功能不全为主要特点的疾病。尽管最近在治疗方面取得了进展,但其发病率以及死亡率仍然很高,目前尚无预防及治疗AKI的特效药物,因此探明其发病机制,找到合适靶点,对AKI的治疗极为重要。Zuk等[41]发现铁死亡在AKI过程中起促进作用。Friedmann等[42]发现GPX4表达缺陷的小鼠两周内会死于急性肾损伤,而减少小鼠体内的脂质可延长小鼠的生存期,根据先前研究推测,GPX4所致的铁死亡可能在小鼠急性肾损伤的发生过程中起了重要作用。15-脂氧合酶(15-lipoxygenase,15-LO)氧化多不饱和磷脂酰乙醇胺(phosphatidylerhanolamine,PE)可实现铁死亡,但15-LO通常以游离多不饱和脂肪酸为底物。Wenzel等[43]发现蛋白激酶抑制剂磷脂酰乙醇胺结合蛋白(phosphatidylerhanolamine binding protein1,PEBP1)与两种15-LO亚型(15-LO-1和15-LO-2)形成复合物,并改变它们的底物以产生氢过氧化物-PE,促进肾细胞的铁死亡。由此可见,PEBP1/15-LO复合物作为铁死亡的主要调节因子对人类疾病有着深远的影响,是开发治疗AKI药物的新靶点。多种小分子物质可以通过调控铁死亡的发生来抑制AKI的发生。Martin-Sanchez等[44]的研究发现,铁死亡经典抑制剂3-氨基-4-环己基氨基苯甲酸乙酯(ferrostain-1,FER1)可抑制叶酸所致的AKI发生,进一步表明了铁死亡在AKI发生过程中的重要性。研究表明,茯苓酸(pachymic acid,PA)可能直接或间接激活NRF2,上调下游铁死亡相关蛋白GPX4、SLC7A11和血红素加氧酶(hemeoxygenase1,HO-1)的表达,抑制铁死亡发生,从而对缺血再灌注所致的小鼠急性肾损伤起保护作用[42],但其具体机制尚未探明,需要进一步试验证实其有效性。Zhang等[45]研究发现,鸢尾素可通过上调GPX4改善由缺血再灌注导致的急性肾损伤,提出了鸢尾素可以通过抑制铁死亡治疗AKI。Hu等[46]的研究表明,维生素D受体可以通过上调GPX4表达水平抑制铁死亡的发生,从而缓解顺铂诱导的AKI。
缺血再灌注损伤是导致AKI的主要原因之一,更好地理解其机制将为 AKI 的预防和治疗提供更多可能。研究表明,铁死亡在缺血再灌注损伤中起关键作用[47]。肝再生增强因子(augmenter of liver regeneration,ALR)是一种广泛分布的多功能生长因子,在包括肾脏在内的所有哺乳动物组织中都有表达,其主要功能是促进肝细胞增殖和肝再生[48]。Huang等[49]发现,在缺血再灌注肾损伤小鼠模型中,ALR可以通过GSH-GPX4调节铁死亡的发生,表明ALR可能是防治缺血再灌注所致的AKI的重要靶点。Ding等[50]研究发现,微小RNA(miRNA,microRNA)也可参与铁死亡的调控。在缺血再灌注肾损伤的模型中,miR182-5p、miR-373-3p表达明显增高,并通过与mRNAGPX4和SLC7A11的3'UTR结合下调GPX4和SLC7A11的表达,从而促进铁死亡的发生。这反映了表观遗传调控修饰在铁死亡中的作用。XJB-5-131是新一代抗氧化剂,具有线粒靶向和自由基清除的双重作用。Zhao等[51]研究证实,XJB-5-131可通过抑制脂质过氧化特异性抑制铁死亡,从而缓解缺血再灌注损伤后的急性肾损伤。该研究体现出XJB-5-131在预防缺血灌注性损伤所致的AKI中的潜力。因此,以诱导铁死亡为目标可能为AKI提供新的预防及治疗手段。但以上研究不足之处在于其多基于啮齿类动物模型,未来可进行体外、哺乳动物以及临床试验,从而将研究结果应用于临床。
4 总结与展望
铁死亡是一种可通过多种机制调控的,以铁离子催化的、脂质过氧化为特征的程序性死亡形式。目前关于铁死亡与肾脏疾病的研究主要集中在肾癌以及AKI上,尚无关于慢性肾脏病(chronic kidney disease,CKD)的研究。大量研究证明,铁死亡在慢性病(如冠心病、阿尔茨海默症)中起重要作用[1],提示铁死亡在CKD中的研究具有广泛前景。在表观遗传修饰调控铁死亡方面,目前仅知道少数的微小RNA可调控铁死亡的发生[52],但其他非编RNA在调控中的作用尚无研究。此外,目前发现了大量小分子药物可用于铁死亡的诱导与抑制,然而这些药物并未转化为临床应用使患者受益。我们认为,接下来的研究可以着手于以下几个方面:1)对铁死亡的发生机制进一步阐明,寻找更多针对铁死亡重要靶点的调节剂;2)继续探究铁死亡调控机制与肾脏相关疾病的关系,研究铁死亡在慢性肾脏病中的作用;3)探索表观遗传修饰介导的铁死亡调控;4)转化目前可用于调控铁死亡的小分子药物,将其用于临床。
该综述阐述了铁死亡的发生机制与其在肾脏相关疾病的研究进展,为肾脏相关疾病的治疗、诊断以及相关研究提供了坚实的基础。
猜你喜欢
黑龙江大学自然科学学报(2022年4期)2022-11-17
河北科技师范学院学报(2022年2期)2022-08-26
现代临床医学(2021年6期)2021-11-20
中成药(2018年9期)2018-10-09
中成药(2018年1期)2018-02-02
中成药(2017年12期)2018-01-19
中成药(2017年4期)2017-05-17
中国塑料(2016年3期)2016-06-15
华东理工大学学报(自然科学版)(2015年4期)2015-12-01
中国洗涤用品工业(2015年4期)2015-02-28
