
基于改进RankComp的个体化差异表达基因识别算法
2021-09-09 00:57:20杨书新陈良泊
江西理工大学学报 2021年4期
杨书新, 陈良泊
(江西理工大学信息工程学院,江西 赣州341000)
0 引 言
基因的选择性表达与生物的生命调控息息相关,研究疾病样本中基因的差异表达有助于揭示疾病相关的生物学机制[1]。现有的许多算法如:T-test[2]、SAM[3]、edgeR[4]和DEseq[5]被提 出 用 于 在 群 体 层 面 上鉴别不同病理生理条件或不同实验条件下的差异表达基因(Differential Expression Gene,DEG)。由于群体水平的差异表达基因忽略了疾病的异质性,不能提供患者特异性的差异表达信息,故而无法回答许多具有生物学意义的问题[6]。例如,Tomlins等发现一些基因只在特定一些疾病样本而不是在所有的疾病样本中出现失调[7]。无独有偶,Zhou等在乳腺癌差异表达研究中发现,在雌激素受体阳性的乳腺癌样本中,一部分差异表达基因的失调方向与其在雌激素受体阴性的乳腺癌样本中的失调方向相反[8]。在个体层面识别差异表达基因是实现精准医疗的基本步骤,有助于更好地理解疾病特异性的生物学机制并促进个性化治疗策略的发展[9]。随着精准医疗概念的普及,研究热点逐渐转向个体化基因差异表达分析。离群度(Outlying Degree,OD)[10-11]、Zscore和其改进Rscore[12]等算法被提出用于识别个体化差异表达基因。不过这些算法对批次效应很敏感,也缺乏统计学控制。为解决该问题,Wang等提出基于相对表达丰度排秩(Relative Expression Orderings,REO)的RankComp算法[13]。RankComp通过比较每个疾病样本中基因对的REO和正常样本中REO高度稳定的基因对来识别个体水平的差异表达基因。由于没有直接使用基因表达值,RankComp独立于批次效应。经研究发现,RankComp存在统计效能低的问题。Richard等提出另一种基于相对表达丰度排秩的算法PenDA[14],声称在个体化基因差异表达分析中效率很高。但是PenDA的结果依赖于两个人工参数,缺乏统计学控制。同时本文研究发现,PenDA在数据清洗过程中会删除潜在差异表达基因,造成生物信息丢失。
为解决以上问题,本文对RankComp算法进行改进,提出RankComp+算法,并在两个仿真数据实验和一个真实的肺癌基因表达数据集实验中检测个体化差异表达基因,评估了RankComp+的性能。结果表明,RankComp+仿真数据实验和真实肺癌配对样本数据中的表现都优于RankComp和PenDA。此外,为进一步评估RankComp+算法得出结果的可靠性,本文将RankComp+算法应用于数据具有单边性的缺血性心肌病(Ischemic Cardiomyopathy,ICM)数据中,对得到的差异表达基因进行功能富集分析,得到了与ICM相关的一些新的生物学通路,验证了RankComp+算法得出的结果具有生物学意义。
1 相关定义
1.1 个体化差异表达基因
差异表达基因一般来说有两个定义。其一,若一个给定的基因i由于处于不同条件下(如环境压力、时间等)表达水平出现显著性差异,则将基因i定义为差异表达基因。其二,在不同条件因素下,因基因突变或者甲基化等结构发生变化而导致差异的基因被定义为差异表达基因。在一般情况下,使用较多的是第一种定义,例如,基因i在正常样本中表达量为GiN,经观察发现其在疾病样本中的表达量为GiD,若经过实验验证,发现GiN和GiD之间具有显著差异,则认为基因i发生了差异表达并将其定义为差异表达基因。
1.2 相对表达丰度排秩
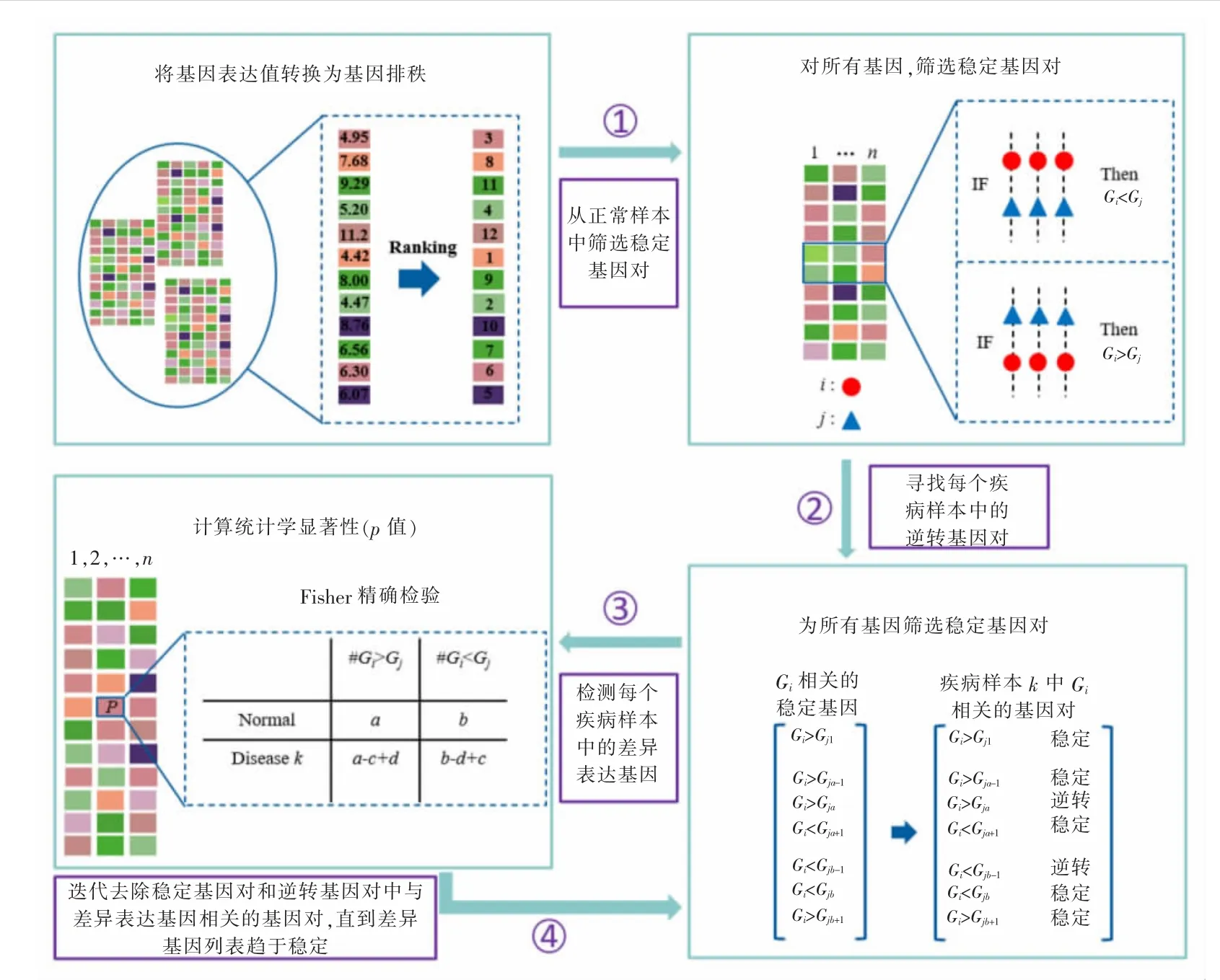
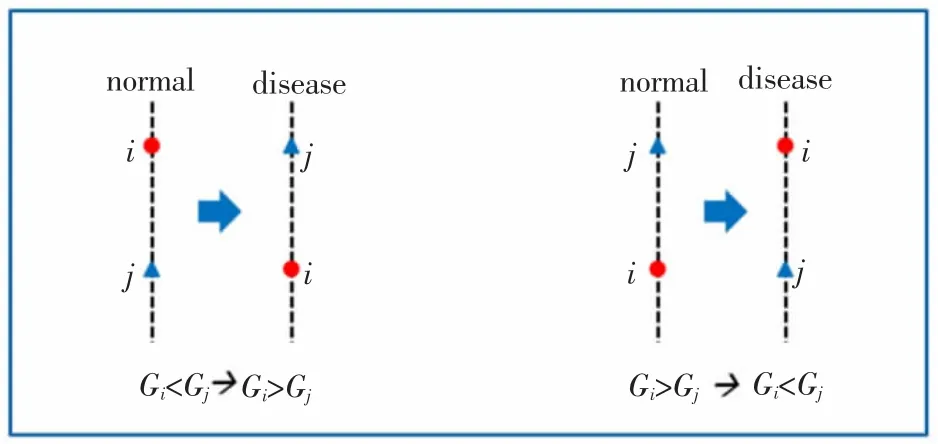
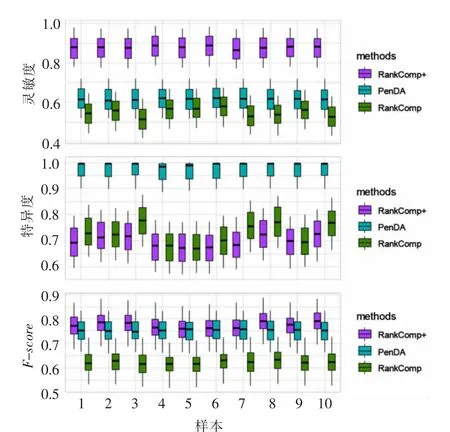
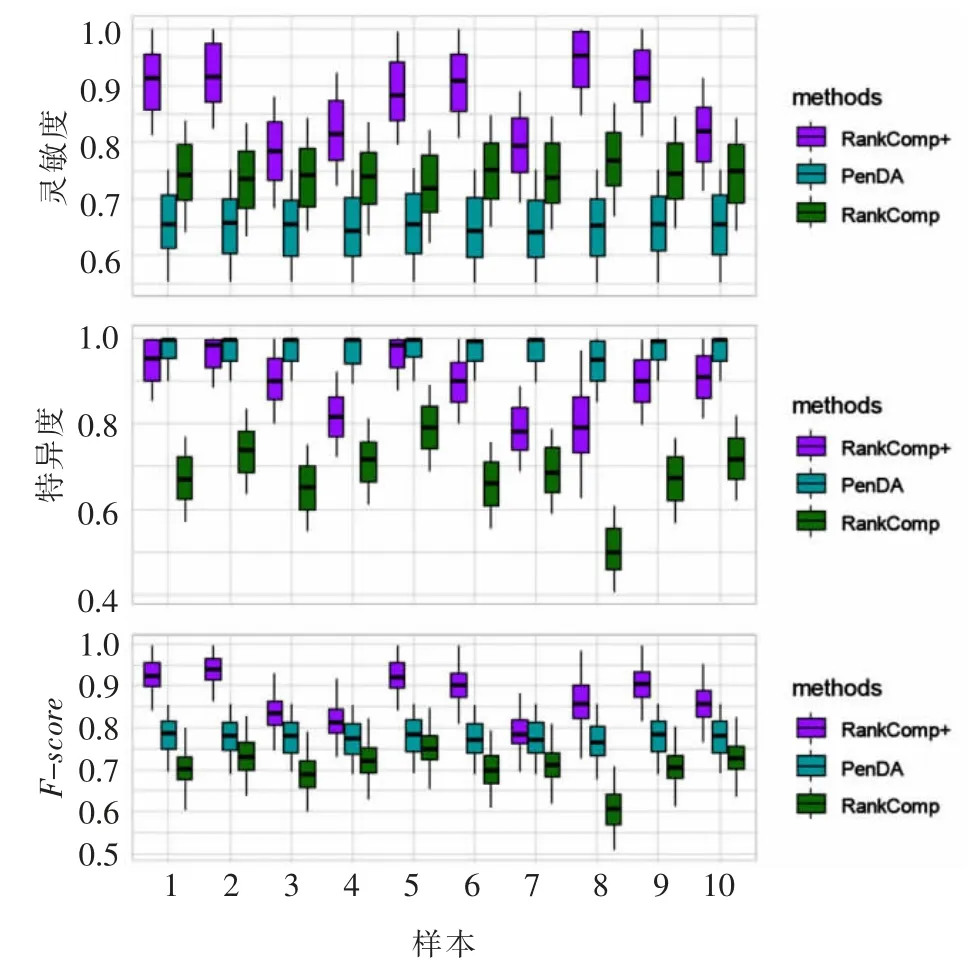
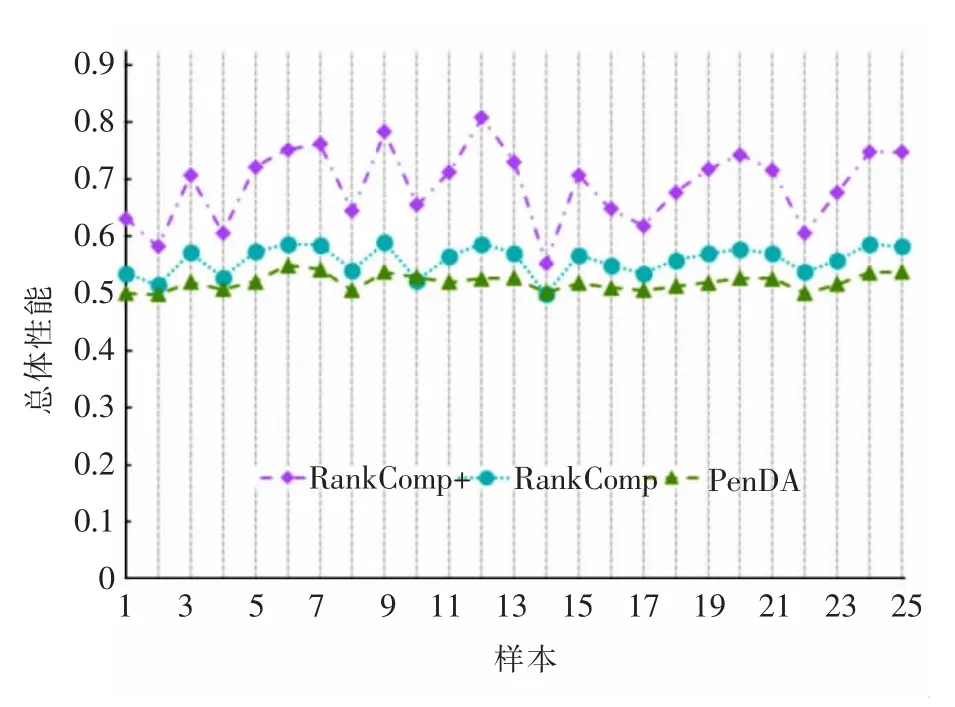
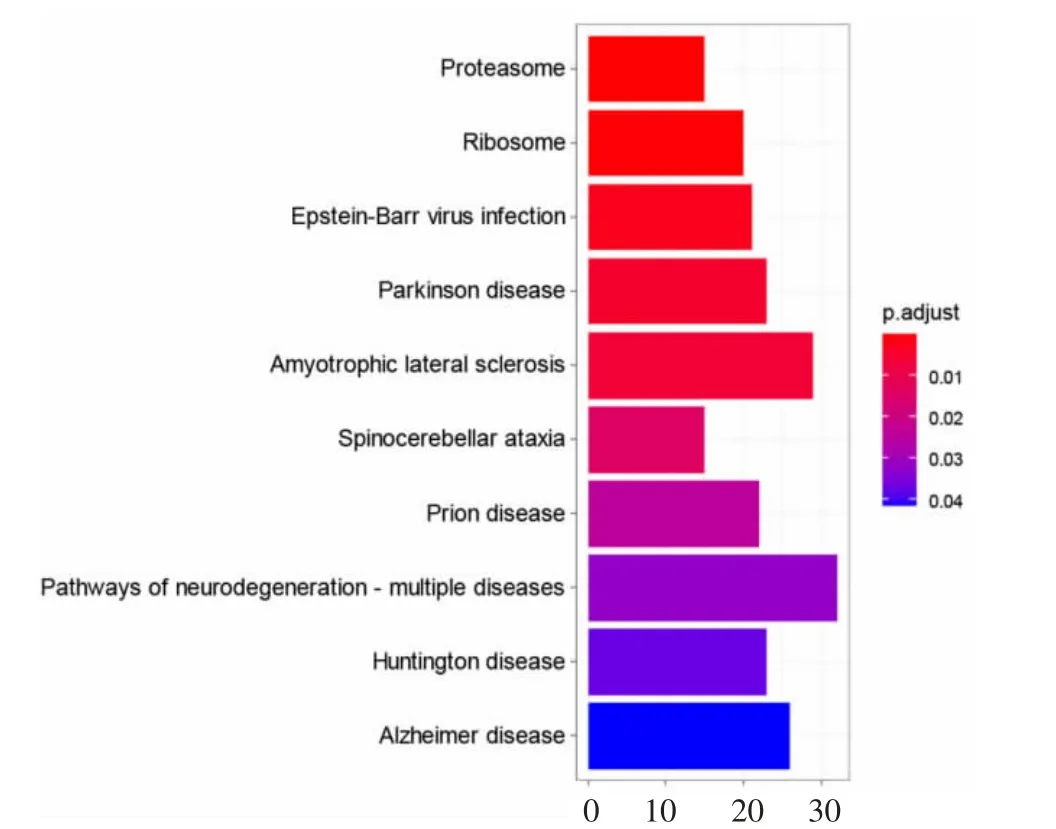
相对表达丰度排秩 (Relative Expression Orderings,REO)是存在于基因对中两基因之间的一种相互关系,由Wang等[13]首先提出。给定一个基因对p包含基因i和基因j,则p的REO有且仅有两种可能,Gi>Gj以及Gi 图1 相对表达丰度排秩示意 为提高RankComp算法的整体效能,RankComp+算法对其进行了两处改进。其一,在稳定对筛选部分,对高度稳定策略的阈值选取进行了调整;其二,在消除伙伴基因差异表达的影响部分,增加了迭代过程,从而使伙伴基因差异表达的影响消除更彻底。RankComp+算法可分为四个步骤,其算法流程图如图2所示。 图2 RankComp+算法差异表达基因识别流程 其中,n表示样本总数;I(x)表示满足x条件的基因对个数。RankComp算法在筛选稳定对时使用高度稳定策略,将阈值设置为99%。本文研究认为该阈值设置过于严苛,容易造成算法统计效能低下的问题。在对高度稳定95%、高度稳定99%和累计二项分布三种稳定对阈值实验验证后(详见3.4节),本文将稳定对阈值定为高度稳定95%。 步骤二,根据筛选出来的稳定基因对,在给定的疾病样本中找出逆转基因对。逆转基因对定义为与稳定基因对中的REO相比,在疾病样本中REO发生改变的基因对。给定基因i、基因j,根据REO的两种不同情况Gi>Gj和Gi 图3 两种逆转对模式示意 步骤三,差异表达基因的判定。差异表达基因的判定原理为,当一个基因对的REO发生逆转时,两个基因至少有一个发生了差异表达。根据该原理,如果控制与待判定基因的伙伴基因的失调对待判定基因的影响,便可以通过基因对的REO来判断待判定基因是否发生差异表达。对于基因i,在控制其伙伴基因j(i≠j)情况下,逆转模式Gi>Gj→Gi 在稳定基因对中,统计REO模式为Gi>Gj的基因对数目,记为a;REO模式为Gi 表1 差异表达判定四格表 设置零假设为a/b=(a-c+d)/(b-d+c),使用Fisher精确检验对表1所列四格表进行检验,得到p值。若p值小于0.05,则说明支持i差异表达上调的基因对于支持基因i差异表达下调的基因对数目有显著差异。通过支持基因i差异表达上、下调的数目c与d的大小即可判定基因i的差异表达方向。若c>d,说明基因i差异表达下调,反之若c 步骤四,伙伴基因差异表达影响的消除。RankComp算法在所有的基因都经过一次Fisher精确检验之后,为了消除伙伴基因的差异表达对待判定基因的影响,从稳定基因对和逆转基因对中去除第一次Fisher精确检验中被判定为差异表达基因的相关基因对,取出后再次执行Fisher精确检验,以第二次筛选出来的DEG列表作为最终DEG。 本文研究认为,RankComp算法对于伙伴基因差异表达的影响消除不够彻底,第二次筛选出来的DEG相关基因对仍然存在于稳定对、逆转对中。为彻底消除伙伴基因差异表达的影响,RankComp+算法在RankComp算法基础上,添加迭代过程。每次得出DEG列表,都去除稳定对、逆转对中与这些DEG相关的基因对,直到前一次和后一次得到的DEG列表一致,才将最后得到的DEG列表作为最后DEG结果。 党支部制定了驿站管理制度、服务群众工作制度、坐班工作制度等,安排机关正式党员作为工作人员到驿站开展服务,先后开展了入户帮扶度夏收、环卫一线送清凉、湿地公园除杂草、城管执法体验日等主题活动,拉近了群众和党员的距离,让群众时刻感受到党组织、党员就在身边。 本研究中使用的所有数据集均从Gene Expression Omnibus(GEO)[15]下载。如表2所列,收集到的肺癌组织的正常样本用来筛选正常条件下具有稳定REO的基因对。其中,GSE27262包含来自同一患者的配对癌症-正常样本的表达数据,本文选择从该数据集中配对样本分析的基因失调方向作为金标准用于在肺癌数据验证实验中的性能评估。本文数据的产生平台主要有两种,Affymetrix平台和Illumina微阵列平台,不同平台的数据预处理方式不同。对于使用Affymetrix平台(HG-U133 Plus2.0)生成的微阵列数据集,原始数据(.CEL文件)从GEO获取并使用RMA算法进行处理。对于Illumina微阵列平台,由于预处理流程相较Affymetrix平台复杂且缺乏比较好的开源分析工具,本文直接从GEO获取处理后的数据。 表2 方法验证实验数据 通过将RankComp+应用于两个仿真实验和一个配对样本实验中,对RankComp+的性能进行了评估。为了使对比效果更为客观,仿真实验采取两种不同策略,其一参考了RankComp算法中设计的仿真策略,其二则采用了PenDA算法中设计的仿真策略。 两种仿真实验均基于正常的肺部数据,重复执行1 000次以避免相对误差。在仿真实验中,灵敏度、特异度和F-score被用来评估方法的性能。灵敏度计算为所有差异表达基因中正确识别的差异表达基因的比例,而特异度则是所有非差异表达基因中正确识别的非差异表达基因的比例。F-score得分是通过灵敏度和特异度如公式(3)计算的统计量。 由于在肺组织配对样本数据中确切的差异表达基因无法获得,本文将成对样本中观察到的失调方向变化作为金标准。通过正确识别的DEG数目与识别的DEG总数的比值计算出的一致性评分能够反映算法识别DEG的准确性。而识别出的DEG数量与基因半数的比值反映算法的统计效能。为综合考虑准确性与统计效能,本文提出总体性能(Overall Power,OP)指标对RankComp+、PenDA和RankComp在肺癌配对样本数据中的表现进行评价,OP的计算方式如式(4)、式(5)所示。 其中,k指的是某一疾病样本;RDEG(k)表示在样本k中别出的DEG数量与基因总数的比值;Consistency(k)表示算法在样本k中的一致性评分。 为确定适合的RankComp+稳定对阈值,本文对常用的高度稳定策略95%、高度稳定策略99%以及累计二项分布策略三种稳定对阈值在配对样本数据集GSE27262中进行了验证,验证结果如表3所列。 表3 RankComp+稳定对阈值选取实验结果 由表3可看出,在高度稳定99%策略下RankComp+算法的一致性指数很高,但是找出的差异表达基因数目很少;而累计二项分布则恰恰相反,找出的差异表达基因数目很多,一致性指数却十分低。为平衡算法的准确性和统计效能,使得算法既能兼顾“找准”又能兼顾“找全”,本文使用高度稳定95%作为RankComp+的稳定对阈值。 本研究在两个仿真实验中评估了RankComp+在个体化差异表达基因识别中的性能。在RankComp策略仿真实验中,首先随机提取GSE19804的10个正常样本作为初始样本。然后,通过随机设置在-2和2之间的倍数变化(log2FC)对其表达值进行改变来随机生成上调和下调的基因。本文通过RankComp策略仿真生成了两个数据集,一个具有3 000个上调基因和3 000个下调基因,另一个具有1 500个上调基因和1 500个下调基因。PenDA策略仿真实验是使用R语言penda包中的方法penda::complex_simulation生成,随机抽取GSE19804的10个正常样本和10个肿瘤样本作为输入数据。生成参数根据penda包的说明设置为size_grp=10,quant=0.05。两次仿真都进行了1 000次以减少相对误差。为了更好地比较它们的性能,本文将RankComp和PenDA也应用在仿真实验中。 在RankComp策略仿真实验中,207个具有20 572个基因的肺组织正常样本被用来识别稳定基因对,155 273 015个稳定基因对被RankComp+筛选出来。同时,由于筛选稳定对的阈值不同,RankComp寻找出133 582 941个稳定基因对用于其DEG检测。各算法在RankComp策略仿真实验中的结果如图4、图5所示。 图4 生成6 000差异基因的RankComp策略仿真实验结果 图5 生成3 000差异基因的RankComp策略仿真实验结果 图4 展示了4种算法在具有3 000上调以及3 000下调的RankComp策略仿真数据集中的表现,从中可以看出RankComp+的表现(F-score平均0.776)优于其他两种方法,由于PenDA算法在执行数据清洗的过程中排除的基因包含仿真出来的差异表达基因,因此PenDA在模拟中显示出较低灵敏度(平均0.618),尽管PenDA在特异度方面表现很好(平均0.971),但低灵敏度导致其F-score较低(平均0.73)。RankComp的表现在某种程度上类似于PenDA,其特异度为平均0.728,而灵敏度平均只有0.550,这导致其F-score较低(平均0.623)。在具有1 500上调基因和1 500下调基因的RankComp策略仿真实验中也可以看到类似的结果(图5)。 对于PenDA策略仿真实验,本文在总共17 816个基因中对10个疾病样本平均仿真了3 122个DEG。使用与RankComp策略仿真实验一样的207个肺组织正常样本,RankComp+提取了109 163 986个稳定基因对,而RankComp获得了90 366 703个稳定基因对作为其DEG检测的背景列表。各算法在PenDA策略仿真实验中的表现如图6所示。 从图6中可以看出,在这些算法中,RankComp的F-score最高(平均0.867),尽管其特异度低于PenDA的特异度(平均0.918 vs.平均0.963),但其较高的灵敏度(平均0.824)导致其F-score比PenDA更好(平均0.867 vs.平均0.857)。值得注意的是,包括RankComp在内的所有算法在PenDA策略仿真实验中均显示出高特异度。与RankComp策略仿真实验相似,RankComp的灵敏度(平均0.514)较差,导致其F-score很低(平均0.668)。 图6 PenDA策略仿真实验结果 总体而言,RankComp+在RankComp策略仿真实验和PenDA策略仿真实验中的总体表现良好,优于其余算法。 为了进一步研究算法的效能,RankComp+、RankComp和PenDA被应用肺癌配对样本数据集GSE27262中。表2中描述的肺组织正常样本被用于筛选稳定基因对。值得注意的是,为了排除GSE27262数据集中正常样本对于结果判定的影响,GSE27262中的正常样本不纳入稳定对选择步骤。实验结果如图7所示。 图7 肺癌配对样本数据集中的表现 肺癌配对样本实验结果显示,在所有参与比较的方法中,PenDA的一致性最高(平均0.993),但其DEG数最低(平均491.16),表明尽管PenDA方法虽然具有较高的准确性,其统计效能却很低,这导致PenDA算法的总体效能很低。RankComp方法也缺乏统计效能,其平均DEG平均数目仅为1 485。RankComp+的一致性得分虽然略低于RankComp和PenDA(平均0.935vs.0.973,0.935vs.0.993),但DEG数却高得多,达到平均4 597.36,致使其总体效能高于RankComp、PenDA算法,表明对RankComp+算法而言,准确性和统计效能达到了很好的平衡。 缺血性心肌病(Ischemic Cardiomyopathy,ICM)是向心脏供血的冠状动脉变窄而引起的一类心肌病,是心脏猝死的主要原因之一。ICM发病率的逐年增加使其对人类健康造成的危害更加严重。缺血性心肌病的数据属于一类“单边数据”,由于疾病特性影响,几乎不太可能获取到同一个体的配对样本,这导致需要依赖配对样本进行分析的各种差异基因识别方法无法应用于缺血性心肌病的差异表达分析。本文将RankComp+算法应用于ICM数据集,通过对两方法检测出的差异表达基因的功能富集分析,寻找可能与ICM相关的生物学通路。 从RankComp+结果中提取的DEG的数目为659。通过对这659个DEG的富集分析,可能与ICM相关的3条GO通路和10条KEGG通路被识别出来。GO和KEGG分析结果分别如图8、图9所示。 图8 Rank Comp+算法GO通路富集结果 图9 RankComp+算法KEGG通路富集结果 GO分析显示这659个DEG显著富集于structural constituent of ribosome、S100 protein binding及cadherin binding通路,显著性水平p<0.05。同时,这些DEG在KEGG富集分析中富集到Proteasome、Ribosome、Epstein-Barr virus infection、Amyotrophic lateral sclerosis、Spinocerebellar ataxia等通路。值得注意的是在GO通路富集和KEGG通路富集分析中均发现与核糖体(ribosome)相关的通路被富集,核糖体相关通路可能与ICM的发病有较高相关性。 本文针对现有个体化差异表达基因识别算法存在的统计效能不足等问题,通过对RankComp算法进行改进,提出RankComp+算法。在两种不同策略的仿真实验和肺癌配对样本实验中的实验结果表明RankComp+算法能够稳定地找出疾病样本中的差异表达基因。在进一步将RankComp+算法应用于缺血性心肌病数据的实验中,通过对RankComp+算法识别的差异表达基因进行通路富集分析,与缺血性心肌病相关的新生物学通路被识别出来,说明RankComp+算法识别的差异表达基因具有生物学意义。综上所述,RankComp+算法能够有效应用于个体化基因差异表达分析并为疾病的机理研究提供新的信息。
2 RankComp+算法
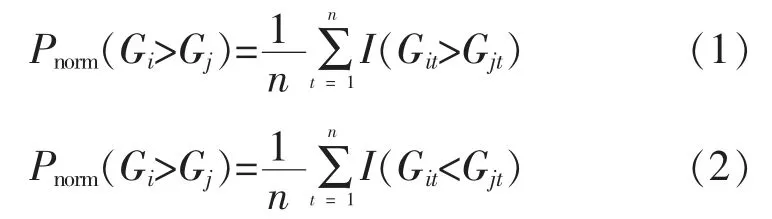
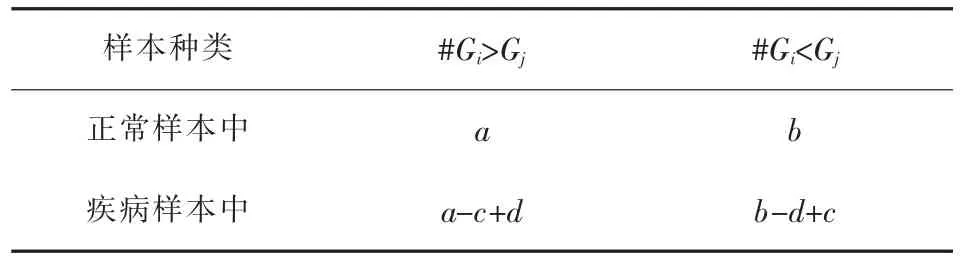
3 实验与分析
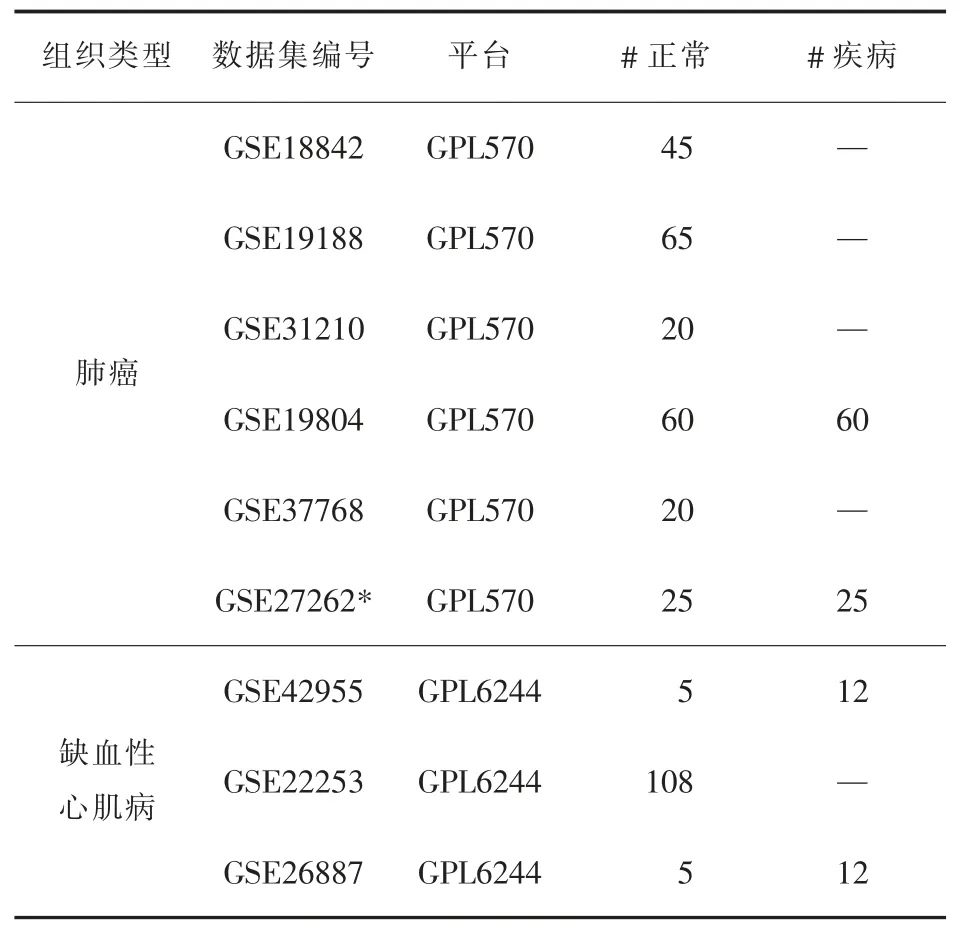
3.1 实验数据
3.2 算法评价指标
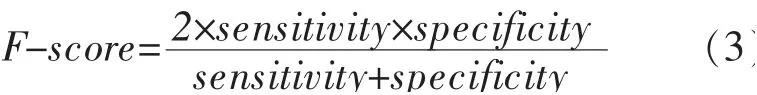
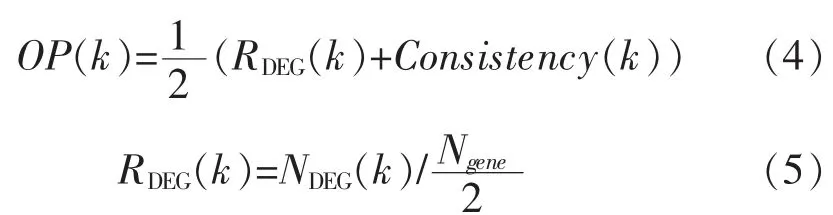
3.3 算法参数确定

3.4 仿真数据实验

3.5 肺癌配对样本数据实验
3.6 缺血性心肌病数据实验

4 结 论
猜你喜欢
中学生数理化·中考版(2022年8期)2022-06-14 06:55:54中国医学影像学杂志(2021年6期)2021-08-13 08:43:38今日农业(2020年22期)2020-12-14 16:45:58制造技术与机床(2019年9期)2019-09-10 07:36:54西南交通大学学报(2018年6期)2018-12-18 02:22:28河北遥感(2017年2期)2017-08-07 14:49:00衡阳师范学院学报(2016年3期)2016-07-10 07:16:27中国病理生理杂志(2015年8期)2015-12-21 12:38:06医学研究杂志(2015年3期)2015-06-10 06:41:52当代经济(2015年4期)2015-04-16 05:57:04
