
肝癌代谢重编程相关基因富集分析及ATP柠檬酸裂解酶表达的临床意义
2021-09-07 13:28周青青李军建郑亦胡余正平朱千东
肝胆胰外科杂志 2021年8期
周青青,李军建,郑亦胡,余正平,朱千东
(温州医科大学附属第一医院,浙江 温州 325000,1.手术室,2.肝胆胰外科)
我国是肝细胞癌(hepatocellular carcinoma,HCC,简称肝癌)的高发国家。在HCC病程中,抑癌基因失活、原癌基因激活及基因突变效应积累等都对HCC发生、发展具有显著诱发和促进作用。最新研究表明,调控肿瘤形成与进展的一个关键因素是肿瘤细胞代谢重编程(metabolic reprogramming)[1],该过程重新界定了肿瘤细胞代谢网络内营养物质的通量与流向,用以满足肿瘤细胞物质代谢和能量代谢,保证肿瘤细胞的存活与增殖,在不利条件下保持生存优势[2]。这些代谢改变为细胞提供核酸、蛋白质等大分子合成所需要的中间代谢物,对于肿瘤的发生和进展至关重要。因此,系统性地研究代谢重编程相关基因表达在HCC预后和治疗中的靶标作用意义重大。在本研究中,我们首先分析了来源于TCGA(The Cancer Genome Atlas)的公共大样本数据,通过富集分析筛选获得参与HCC肿瘤进展的代谢重编程相关基因;其次,在候选基因中ATP柠檬酸裂解酶(ATP citrate lyase,ACLY)作为肿瘤细胞脂质代谢通路上游的关键酶,在许多肿瘤的发生和进展中具有重要作用,因此我们在HCC临床标本中检测ACLY表达水平,并纳入GEO(gene expression omnibus)的大样本表达数据和临床数据进一步研究,明确ACLY表达在HCC生存预后中的作用。
1 材料和方法
1.1 材料
1.1.1 TCGA数据:下载数据库中所有374例HCC测序数据(截止2019年9月),将其中50对HCC与癌旁配对样本纳入差异表达分析。标本以编号区分肿瘤组织或癌旁组织,基因表达数据为level 3级别。该测序数据完成于Illumina HiSeq 2000测序平台。
1.1.2 GEO数据:下载数据库中基因芯片数据集GSE14520,该数据集包括247 例HCC基因芯片数据和临床数据。本研究排除26 例样本:其中22 例样本隶属GPL571 平台,5 例无临床结果数据(1 例隶属GPL571 平台,并且无结果数据)。最后,隶属GPL3921 平台的221 例基因芯片数据被纳入最终分析。所有肝组织标本均来自2002年至2003年在复旦大学附属肝癌研究所和中山医院接受根治性切除的患者[3-4],随访数据和术前检查数据较完整。
1.1.3 人HCC组织标本的选取:组织标本和癌旁组织标本来自温州医科大学附属第一医院2018年5月至10 月间因原发性HCC行肝切除术的15 例病例的HCC组织标本及癌旁组织,其中男11 例,女4 例,年龄37~78岁,平均55.8岁。术前未接受放疗、化疗等治疗,术中均未见远处转移,术后经病理证实为HCC,其中高分化7例,中分化6例,低分化2例。所有患者均签署知情同意书,本研究经温州医科大学附属第一医院医学伦理委员会批准。
1.2 研究方法
1.2.1 基因富集分析:本研究采用的基因富集方法包括GSEA(gene set enrichment analysis)、GO(gene ontology)和KEGG(Kyoto encyclopedia of genes and genomes)。GSEA无需先找差异基因,计算的基本原理是扫描排序序列,当出现一个特定功能基因集中的基因时,就增加该功能集富集评分(enrichment score,ES)值,反之,就减少ES值,所以在整个扫描过程中,ES是一个动态的值,可以避免一些人为筛选差异基因导致的偏倚。GO可分为分子功能(molecular function)、生物学过程(biological process)和细胞组分(cellular component)三个部分,分别对基因进行注释和分类。我们通过应用Cytoscape软件和BiNGO插件进行分析[5-6]。KEGG通路富集分析以KEGG通路为单位,应用统计学检验,在整个基因组背景中找出差异表达基因中显著性富集的通路。此分析同样需要筛选出候选的差异基因列表。在本研究中,我们通过在线网络工具KOBAS[7](http://kobas.cbi.pku.edu.cn/)进行此分析。
1.2.2 样本总RNA提取及qRT-PCR检测:按照Invitrogen公司Trizol试剂盒说明书进行操作,提取总RNA,并用琼脂糖凝胶电泳判断RNA有无降解,检测所提取RNA的浓度和纯度,RNA的A260/A280在1.8~2.0 范围内者方可进行后续实验。根据反转录试剂盒说明书将其反转录合成cDNA,避光保存于-20 ℃冰箱。以此cDNA为模板,GAPDH为内参,荧光定量PCR反应使用SYBR法Q-PCR试剂盒在BIORAD CFX96 荧光定量PCR仪上操作。PCR反应条件:94 ℃ 30 s;94 ℃ 5 s;55 ℃ 30 s,共35个循环。最后采用2-△△Ct方法计算基因相对表达值。引物委托上海生物工程公司合成,序列如下:ACLY(F’TC GGCCAAGGCAATTTCAGAG,R’CGAGCATACTT GAACCGATTCT);GAPDH(F’GTCTTCACCACCA TGGAGAAG,R’CAAAGTTGTCATGGATGACCTT GG)。
1.3 统计学分析
TCGA RNAseq数据通过R软件(http://www.rproject.org/,version 3.2.5),采用R包“edgeR”包来构建表达矩阵,评估组内和组间差异,进行差异表达基因分析,获得差异表达基因列表;基因分析纳入标准raw read counts>1,差异表达基因筛选标准为:错误发现率(false discovery rate,FDR)<0.05,log CPM>1 且|差异倍数(fold change,FC)|>3。GEO数据分析首先根据ACLY表达值将样本分为高表达组和低表达组,生存分析和Cox比例风险模型分析过程采用R包“survival”;后者先进行单因素Cox回归分析,再将差异显著的进行多因素Cox回归分析。其他数据统计分析采用SPSS23.0统计软件进行。两组数据间统计差异的比较采用独立样本t检验;各个临床因素与ACLY表达值组成差异采用χ2检验;P<0.05表示有统计学意义。
2 结果
2.1 GSEA分析获得与HCC代谢重编程相关的差异表达基因
通过构建表达矩阵、基因ID转基因名称等步骤获得全基因表达谱列表,包含27 915 个基因,该列表满足GSEA的分析要求。本研究筛选差异最显著的3条代谢通路进行下一步研究(表1)。
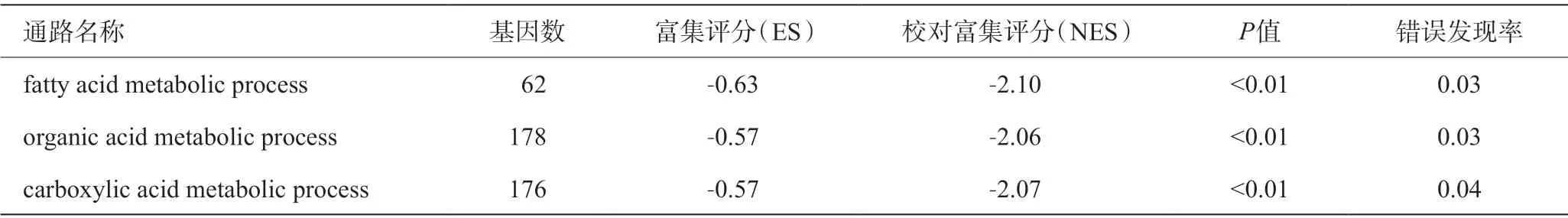
表1 在HCC癌旁组中富集最显著的代谢相关通路
2.2 GO分析获得与HCC代谢重编程相关的差异表达基因
筛选全基因表达谱列表,获得1 720个差异表达基因,其中表达上调的基因1 052个,表达下调的基因668个(图1)。经GO分析,本研究选择差异最显著3个代谢相关生物学过程(表2)纳入后续研究,共包含122个基因。

表2 肿瘤组织和癌旁组织差异最显著的代谢相关生物学过程通路
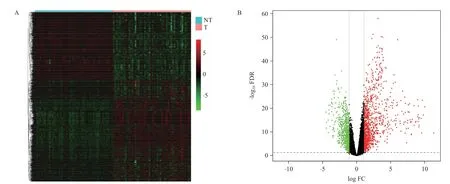
图1 肿瘤组织和癌旁组织差异表达基因的热图(A)和火山图(B)
2.3 KEGG通路分析获得与HCC代谢重编程相关的差异表达基因
在http://kobas.cbi.pku.edu.cn/网站输入差异表达基因列表,参数选择:Databases:KEGG pathway;Statistical test method:hypergeometric test/Fisher’s exact test;FDR correction method:Benjamini and Hochberg,差异最为显著的KEGG通路见表3,其中metabolic pathways包含200个差异表达基因,用于后续研究。
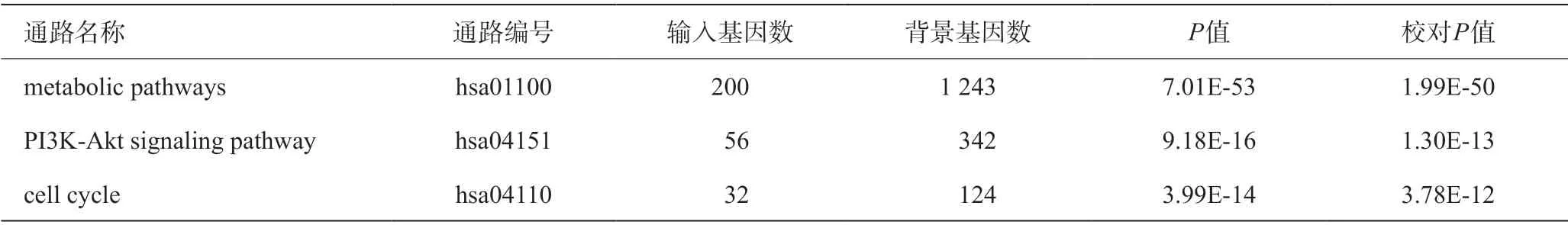
表3 肿瘤组织和癌旁组织差异最显著的KEGG通路
2.4 联合分析筛选获得HCC代谢重编程相关基因
单独应用GSEA、GO或者KEGG分析存在偏倚的可能性,我们将上述3 种富集分析方法获得的代谢重编程相关的差异表达基因取交集,获取共同的代谢重编程相关的差异表达基因,共有26个基因(图2),其中表达上调的差异基因8个,表达下调的基因18个。

图2 筛选获得26个代谢重编程相关基因
2.5 HCC患者肿瘤组织与癌旁组织中ACLY基因表达
为了验证ACLY在HCC临床样本中的表达丰度,我们采用qRT-PCR的方法检测了15对HCC组织和癌旁组织的标本,结果表明,HCC组织ACLY表达值显著高于癌旁组织(P<0.01)(图3)。

图3 qRT-PCR检测15对HCC组织与癌旁组织表达情况(**P<0.01)
2.6 GEO大样本数据生存分析和临床相关性研究验证
为了明确ACLY高表达在HCC患者预后中的作用,我们纳入GEO大样本数据予以验证,该组基线数据较完整(表4)。我们首先提取GEO组表达数据,显示ACLY在HCC组织的表达值高于癌旁组织(P<0.01)(图4)。

图4 HCC组织样本ACLY表达量显著高于癌旁组织(**P<0.01)
根据ACLY表达中位数,将临床HCC样本分为高表达组和低表达组(由于部分数据缺失,每个的临床指标数目略有差异)。临床相关分析因素包括:性别、年龄、乙肝状态、谷丙转氨酶水平(>50 U/L/≤50 U/L)、主瘤直径(>5 cm/≤5 cm)、多发病灶、肝硬化、TNM分期、BCLC分期、CLIP分期、AFP水平(>300 ng/mL/≤300 ng/mL)。分别统计了与ACLY表达相关的因素(表4),及其在HCC生存预后中的作用(图5,表5、6)。
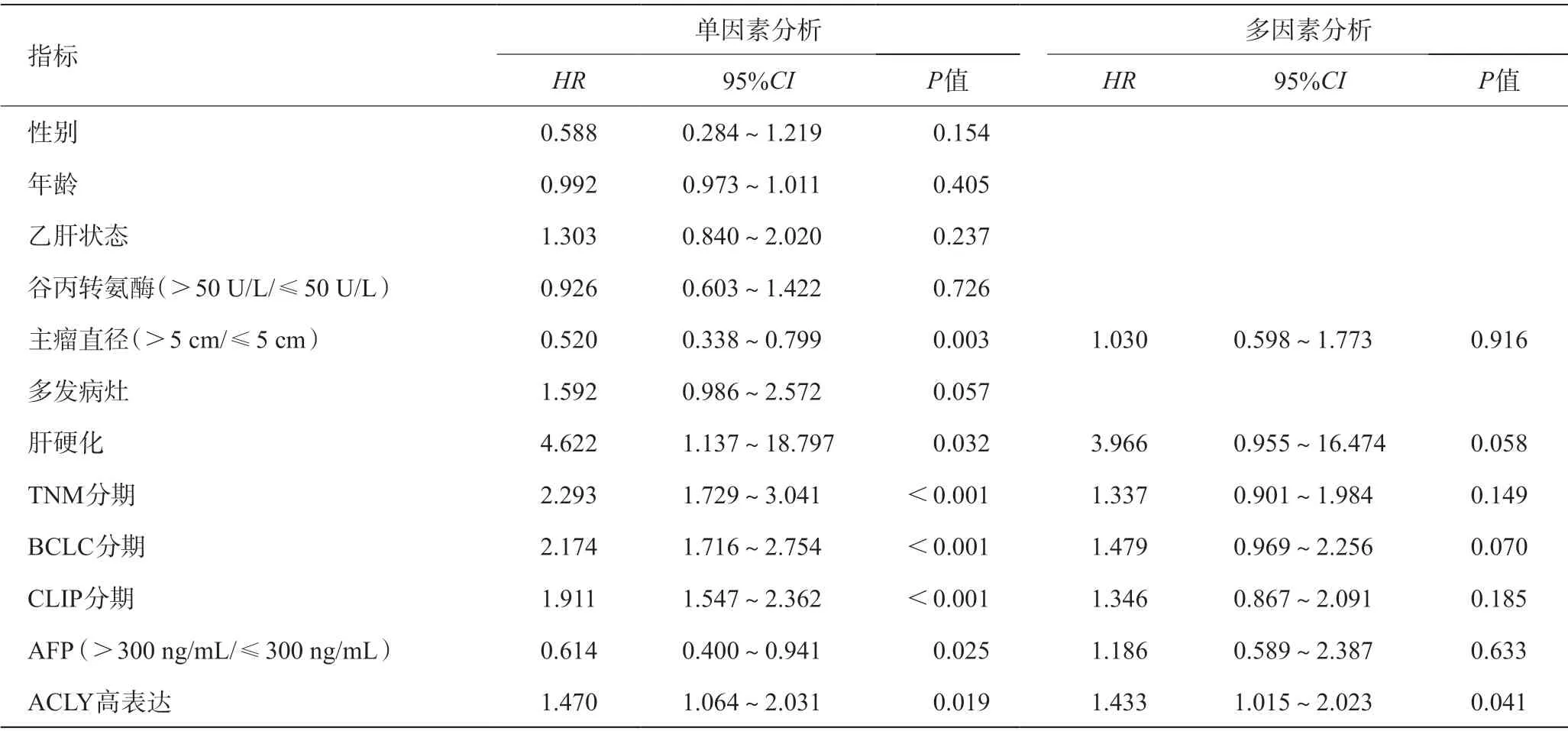
表5 HCC总体生存率Cox回归分析
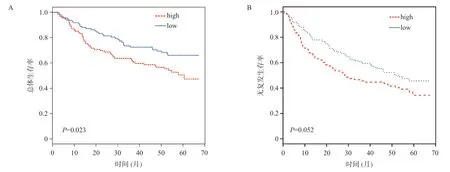
图5 ACLY高表达及低表达组患者总体生存率比较(A)与无复发生存率比较(B)
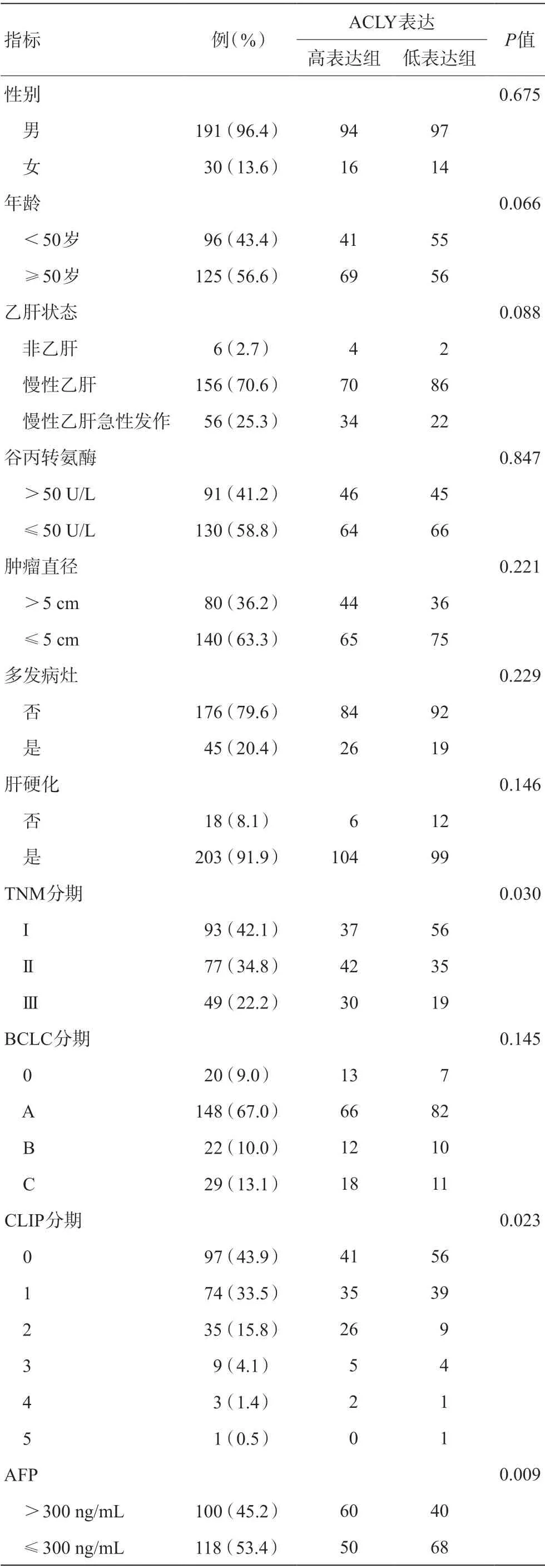
表4 HCC代谢重编程相关基因ACLY表达与临床指标的相关性
3 讨论
HCC是常见的恶性肿瘤,其发病率和病死率在恶性肿瘤中均位居前列[8]。在临床上通常采用病理指标指导肿瘤分期,选择治疗方法和分析患者生存预后,其中肿瘤血管侵犯、分化程度、大小和数目、TNM分期、BCLC分期及CLIP分期等都是影响HCC预后的重要因素[9]。随着分子生物学的进展,相关研究显示HCC的发生和进展实质上是一个动态变化的过程,包括抑癌基因失活、原癌基因激活、基因突变效应积累等[10-11]。因此,许多研究者试图运用特定基因表达变化来进行肿瘤的分型和预后评估,预测肿瘤的发生、复发和转移等。许多癌症相关分子的异常表达与患者预后密切相关,是肿瘤预后的
独立危险因素[12-13],这些分子同样包括了部分代谢相关基因。代谢相关基因在肿瘤细胞中异常表达,广泛参与肿瘤的侵袭、转移等生物学行为,并可能是癌症患者生存预后的指标和治疗靶点。
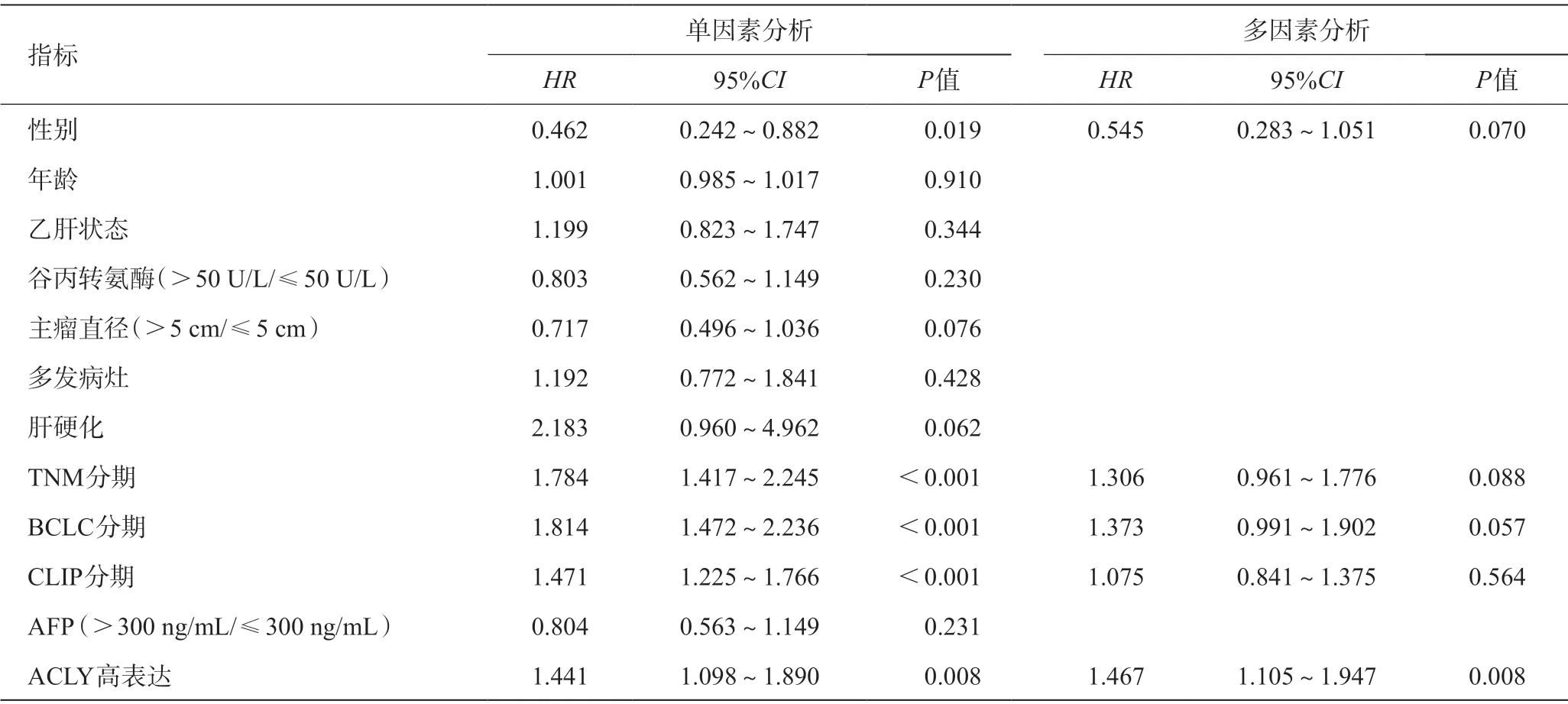
表6 HCC无复发生存率Cox回归分析
目前,已有大量分析肿瘤差异表达基因的高水平研究发表,几乎涵盖了所有肿瘤类型,但是这些研究并不限定于特定的肿瘤表型,尤其是分析HCC代谢重编程相关基因本研究是属首次。另外,在以往类似的研究中,往往通过分析基于基因芯片的表达数据,筛选差异候选分子,难以保证1:1 的配对,从而影响了数据的完整性和可信度。此外,基因芯片等来源的表达数据通常需要进行大规模的qRT-PCR来验证候选分子的表达值。而已有研究报道,高通量测序技术的结果相比于高通量基因芯片结果,定量的可信程度更高,灵敏度更强,可以作为基因表达丰度的定量方法。因而,本研究基于TCGA数据库的大样本生物信息测序数据,筛选HCC差异表达基因列表,并通过差异表达基因的聚类分析,富集代谢相关的信号通路,筛选最有显著价值的共同代谢重编程相关基因。我们下载TCGA数据库内现存所有的374例HCC相关样本的详实数据,其中肿瘤与癌旁配对的50对标本纳入差异表达基因分析。由于总体低表达基因(low read counts)通常不是差异分析的目的基因,对研究结果意义不大;因此,我们只将每个基因平均raw read counts>1 者纳入分析。并设置最终差异表达分子纳入标准为:FDR<0.05,log CPM>1且|FC|>3。在筛选过程中,既往文献报道一般将差异表达的标准设定为|FC|>1.25~2.00,P<0.05,对表达量log CPM没有要求。本研究中设定的差异表达标准筛选出的差异表达分子在标本中既达到一定的表达量,又同时保证了肿瘤组织与癌旁组织表达值之间具有统计学差异,结果的可信度更强,也更加具有临床意义。为了更加精确筛选和全面评估参与HCC代谢重编程的基因,减少应用单一通路富集分析方法存在偏倚的可能性,本研究采用GSEA、GO和KEGG共3种通路富集方法,最终我们筛选了26个代谢重编程相关基因。
ACLY是26 个代谢重编程相关基因之一,是脂质代谢通路上游的关键酶[14],在肿瘤细胞的脂肪酸从头合成中发挥着重要作用,催化内源性脂质从头合成的第一步反应,即线粒体来源的辅酶A和柠檬酸经其催化转变为草酰乙酸及乙酰CoA。在肿瘤的代谢重编程中,脂肪酸合成代谢具有特殊重要的作用[15]。脂肪酸来源可分为外源性吸收和内源性从头合成,大多数正常细胞通过外源性途径获取,然而在肿瘤细胞中超过90%的脂肪酸来源于内源性从头合成[16]。脂肪酸从头合成速率增加可显著促进细胞膜物质的生物合成。脂肪酸合成增加,尤其是从头合成增加,是恶性肿瘤细胞的重要特征之一[17-18]。抑制肿瘤细胞的内源性脂肪酸合成,可诱导肿瘤的退化[19]。ACLY在癌细胞中常见上调表达,在多种肿瘤中,ACLY的活化和表达能够促进肿瘤生长,比如膀胱癌、结直肠癌、胶质母细胞瘤、肺癌等[20-23]。在结直肠癌中,ACLY的过表达可以诱导伊利替康的耐药,但敲减ACLY可以逆转该结果[20]。在许多永生化细胞系中,ACLY异常上调表达也普遍存在[24]。此外,通过抑制ACLY可以阻断MAPK和PI3K/Akt通路,从而抑制癌细胞的生长和增殖[25]。但在HCC中ACLY表达水平尚未见报道。我们的临床标本检测结果和来源于GEO的数据均表明ACLY在HCC组织中的表达水平显著高于癌旁组织,与其他癌症类型类似。
为了进一步明确ACLY过表达在HCC预后中的意义,我们纳入GEO数据进行临床相关性研究。分析结果表明:ACLY的异常表达与TNM分期、CLIP分期、AFP相关(P<0.05);ACLY的异常高表达与HCC患者总生存率相关(P=0.023),而且与无复发生存率存在相关性的趋势(P=0.052)。总体生存单因素Cox回归和多因素Cox回归分析表明,仅ACLY的表达水平与总体生存显著相关(P=0.041)。无复发生存单因素Cox回归和多因素Cox回归分析表明:仅ACLY表达水平与患者术后无复发生存显著相关(P=0.008)。ACLY表达水平与患者预后生存时间具有显著的相关性,可以作为HCC生存预后的一个独立风险预测指标,其效能可能优于常见的临床病理指标。
总体而言,ACLY的表达上调是HCC患者预后的独立危险因素。代谢重编程相关基因的功能相对明确,可作为肿瘤靶向治疗的靶点,逆转代谢重编程过程,进而有助于控制HCC的进展,这可能是今后HCC的有效治疗策略。
猜你喜欢
少先队活动(2021年2期)2021-03-29
中学生数理化·高一版(2021年2期)2021-03-19
汽车维修与保养(2021年8期)2021-02-16
学生天地(2020年17期)2020-08-25
数学大王·低年级(2020年3期)2020-03-12
知识经济·中国直销(2018年8期)2018-08-23
数学学习与研究(2017年3期)2017-03-09
中国老区建设(2016年1期)2016-02-28
中国病理生理杂志(2015年8期)2015-12-21
医学研究杂志(2015年3期)2015-06-10
