
全自动固相萃取-UPLC-MS/MS法检测清咽类保健食品中非法添加的盐酸二氧丙嗪和沙丁胺醇
2021-09-02 06:46吴鸳鸯张丽媛程巧鸳
食品工业科技 2021年16期
王 任,吴鸳鸯,胡 磊,张丽媛,程巧鸳
(浙江省食品药品检验研究院,浙江杭州 310052)
近年来,随着人们自我健康意识的提高以及我国大健康产业迅猛发展,消费者对保健食品的需求进一步增加。一些不法商家为吸引消费者,谋取高额利润,在保健食品中非法添加短期内见效快、副作用大的化学药物[1−5],这种情况多见于清咽类保健食品[6−9]。盐酸二氧丙嗪作为一种抗组胺药,具有镇咳、祛痰、平喘和粘膜表面局麻等作用[7]。沙丁胺醇是选择性β2受体激动剂,可通过松弛气道平滑肌扩张支气管,缓解支气管痉挛[10]。在某些清咽类的保健食品中添加了这两类化学成分[11],以达到快速缓解咽喉不适的效果,但长期食用此保健食品会导致机体依赖并加重病症[12−13],因此需要积极开展对化学药物非法添加检测方法的研究。
由于保健食品基质成分复杂,采用固相萃取净化处理可以达到富集目标物质和净化效果。传统的固相萃取技术是人工操作,会有平行性不好,操作过程时间长等缺点。近年来,在人工固相萃取的基础上改进的全自动固相萃取实现了活化、上样、淋洗、干燥、洗脱全过程依照程序设置自动完成,相对传统的固相萃取装置有节约时间、溶剂和人力成本等优势。全自动化的样品前处理过程不仅可解决传统固相萃取操作耗时的缺点,还可减少人为误差[14−17]。已有文献报道传统的固相萃取方法处理保健食品[17−20],但利用全自动固相萃取技术处理样品的研究却鲜有报道。同时在UPLC-MS/MS检测过程中,基质效应也是不可忽略[21],尤其是针对基质复杂的保健食品,需对样品提取液进行有效的净化才能得到准确的定量结果。通过混合型阳离子交换固相萃取柱(MCX),可以降低UPLC-MS /MS测定过程中的基质效应。
目前,保健食品中非法添加化学物质的检测方法有TLC[22−23]、HPLC[24−26]、HPLC-MS /MS[7,27]、UPLCQTOF MS[28−29]法等方法。薄层色谱法易受干扰导致定性定量不准确,而且容易出现假阳性;高效液相色谱检测结构类似物质时会有干扰,同时检出下限较高[30−31]。而UPLC-MS/MS联用技术能将高效液相色谱对复杂样品的高分离能力与三重四极杆质谱的高灵敏性和高选择性有效结合起来,因此色谱-质谱联用技术具有灵敏度高、稳定性好、定性定量更准确等特点而被广泛用于检测工作。
本研究以清咽类保健食品为研究对象,通过全自动固相萃取技术净化样品,采用超高效液相色谱-串联质谱法同时测定清咽类保健食品中非法添加的沙丁胺醇和盐酸二氧丙嗪,该方法前处理简单、重复性、准确性好,适合大批量样品中非法添加的化学物质的快速测定,以期为保健食品中非法化学物质添加监管提供技术支持。
1 材料与方法
1.1 材料与仪器
保健食品样品 基质包括含片(立铁钻牌铁皮枫斗含片,都乐牌金嗓子喉片)、糖果(京都念慈菴琵琶糖,潘高寿胖大海润喉糖)以及液体(京都念慈菴蜜炼川贝枇杷膏,同仁堂枇杷秋梨膏)3种剂型,国家、省级抽检及市售;甲酸铵、甲酸、甲醇、乙腈 均为色谱纯,Sigma 公司;氨水、乙酸铵 均为分析纯,国药集团;水 超纯水;沙丁胺醇对照品(含量99.7%,批号100204-201103)、盐酸二氧丙嗪对照品(含量99.9%,使用前105℃干燥2 h,批号100299-201202)中国食品药品检定研究院;CQUITY UPLC BEH C18(2.1 mm×50 mm,1.7 μm)、ACQUITY UPLC BEH HILIC-C18(2.1 mm×50 mm,1.7μm)、Oasis HLB固相萃取柱(3cc/60 mg)、Oasis MCX固相萃取柱(3cc/60 mg)Waters;ZORBAX SB-C18(2.1 mm×50 mm,1.8μm) Agilent ;C18固相萃取柱(200 mg) Agela。
1290-G6460QQQ三重四极杆质谱仪 美国安捷伦;Fotector Plus型高通量全自动固相萃取仪 睿科公司;Genpure Pro UV/UF超纯水系统 美国热电公司;MS205DU 电子天平 瑞士梅特勒-托丽多仪器公司;P300H超声波清洗器 德国艾尔玛公司。
1.2 实验方法
1.2.1 标准溶液配制 精确称取沙丁胺醇对照品42.84 mg至100 mL棕色量瓶中,用甲醇溶解,超声10 min(200 W,30 k Hz),放冷,甲醇定容,即得沙丁胺醇储备液。准确称取盐酸二氧丙嗪对照品49.28 mg于100 mL棕色量瓶中,甲醇溶解,超声10 min (200 W,30 kHz),放冷,甲醇定容,即得盐酸二氧丙嗪标准储备液。分别精确移取沙丁胺醇标准储备液和盐酸二氧丙嗪标准储备液各1.0 mL于100 mL棕色量瓶中,用甲醇定容,即得混合标准储备液,置于4℃保存。
1.2.2 供试品溶液制备
1.2.2.1 提取 取一次口服剂量供试品(片剂:立铁钻牌铁皮枫斗含片)(糖果和液体剂型也取相当于一次口服剂量),研细,精确称取0.5 g(精确到0.0001 g),置于50 mL棕色容量瓶中,加 p H5.2的0.2 mol/L乙酸铵缓冲溶液25 mL,超声10 min (200 W,30 kHz),加约20 mL 甲醇溶解,超声10 min (200 W,30 kHz),放冷,甲醇定容[32]。
1.2.2.2 净化 将提取液全部转移至全自动固相萃取仪中进行净化富集,采用MCX固相萃取柱,净化富集程序见表1。收集洗脱液于10 mL试管中,40℃下吹氮至近干,用10%甲醇水溶液(内含0.1%甲酸)溶解残渣,转移定容至50.0 mL容量瓶,得到待测液,待测液经过0.22μm有机滤膜过滤后待上机测定。
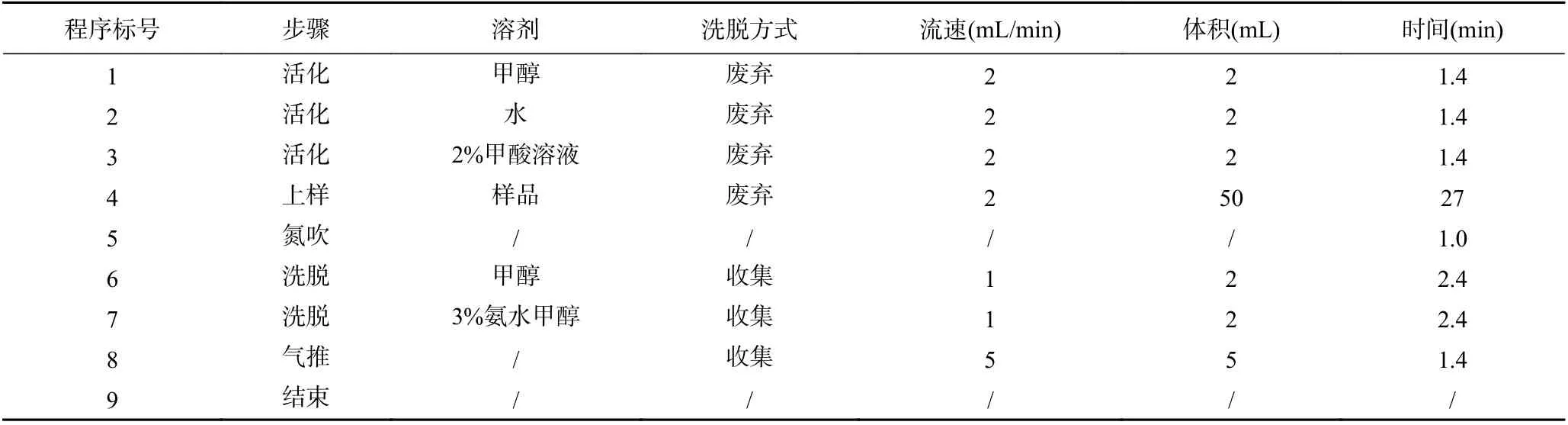
表1 全自动固相萃取仪的净化富集程序Table 1 Program of purification and enrichment by the automatic solid phase extraction installation
1.2.3 阴性空白样品溶液制备 取供试品的阴性空白基质(立铁钻牌铁皮枫斗含片:未检出待测化合物),按“1.2.2”项下条件制备阴性空白样品溶液。
1.2.4 UPLC-MS/MS的色谱条件 采用ACQUITY UPLC BEH C18(2.1 mm×50 mm,1.7μm)色谱柱;柱温:25℃;流速:0.2 mL/min;进样体积:1.0μL;梯度洗脱:流动相A为含有10 mmol/L甲酸铵的0.1%(v/v)甲酸溶液,流动相B为甲醇,流动梯度洗脱程序见表2。

表2 流动相梯度表Table 2 Flow phasegradient table
1.2.5 UPLC-MS/MS的质谱条件 扫描方式:电喷雾离子源(ESI+),正离子模式扫描模式;检测模式:多反应监测(MRM)扫描;毛细管电压:3000 V;离子源温度:300℃;脱鞘气流速:11 L/min,雾化气、气帘气、辅助气和碰撞气均为高纯氮气。优化后相关质谱参数见表3。

表3 质谱MRM参数Table 3 Mass spectrometry parameters
1.3 数据处理
采用QQQ quantitative analysis.B0700和Excel 2003软件进行平均值、回收率及相对标准偏差计算,采用Origin 8.0软件作图。
2 结果与分析
2.1 提取溶剂的选择及优化
沙丁胺醇和盐酸二氧丙嗪均易溶于极性溶剂。本试验探索了不同提取液对待测物提取效率的影响,样品经不同溶剂提取后,经固相萃取柱净化,复溶上样分析。本试验选取100%甲醇、乙酸铵缓冲溶液:甲醇(2:5,v/v)、乙酸铵缓冲溶液:甲醇(5:5,v/v)、乙酸铵缓冲溶液:甲醇(5:2,v/v)进行了比较,结果见图1。待测加标样品过MCX固相萃取柱净化后,测定结果发现100%甲醇提取液提取的待测溶液中沙丁胺醇回收率为0,盐酸二氧丙嗪回收率为98.2%~98.6%,RSD为2.1%~3.0%。乙酸铵缓冲溶液:甲醇(2:5,v/v)提取液提取的待测溶液中沙丁胺醇回收率为88.5%~90.1%,RSD为3.5%~3.9%,盐酸二氧丙嗪回收率达到95%以上,随着乙酸铵缓冲溶液在提取溶液中的比例增加,沙丁胺醇回收率提高并趋于稳定,维持在95%(RSD<5%)以上,而盐酸二氧丙嗪回收率未有太大改变。这是因为沙丁胺醇在乙酸铵缓冲溶液的提取液里带有正电荷,过MCX固相萃取柱后可以与其通过静电作用结合,而在100%甲醇提取液里无法与MCX固相萃取柱结合从而被洗脱下来[33]。同时基于环保与成本考虑,本试验选用乙酸铵缓冲溶液:甲醇(5:5,v/v)作为提取溶液。
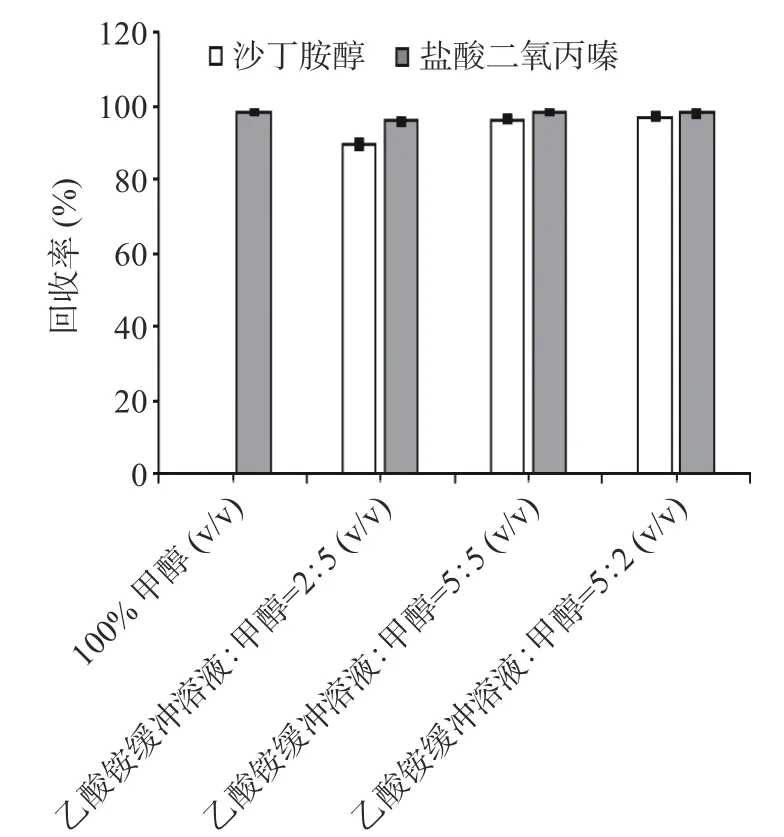
图1 不同提取溶剂及溶剂比例对待测物回收率的影响(n=3)Fig.1 Effect of different extraction solvents and solvent ratio on recoveries(n=3)
2.2 固相萃取柱优化
由于保健品基质较为复杂,其中的糖类、色素等杂质会干扰目标物的检测,在进行前处理时,需要消除这些杂质对目标物的干扰,因此需对提取后的溶液进行净化处理。本试验考察了C18固相萃取柱、HLB固相萃取柱及MCX固相萃取柱对沙丁胺醇和盐酸二氧丙嗪测定结果的影响,结果见图2。结果发现,C18固相萃取柱和HLB固相萃取柱对沙丁胺醇并无保留,盐酸二氧丙嗪的回收率较好,均达90%以上,RSD<5%。而MCX固相萃取柱的使用方法是先用酸化的试剂进行清洗后再用碱化试剂洗脱,溶液中的沙丁胺醇和盐酸二氧丙嗪经MCX固相萃取柱净化后均获得了较高的回收率,且重复性较好,其中待测两种物质的加标回收率均达90%以上,RSD<5%。因为MCX固相萃取柱有效整合了C18固相萃取柱和HLB固相萃取柱的优点,组分中既包含与C18固相萃取柱和HLB固相萃取柱类似的疏水基团,可以通过疏水作用去除提取液中的糖、色素等杂质,又包含强阳离子基团(磺酸基),可通过静电作用力吸附待测的沙丁胺醇,通过淋洗液和洗脱液的合理选择实现保健食品中待测物质的有效净化。因此,试验选用MCX型固相萃取柱对样品进行净化富集。
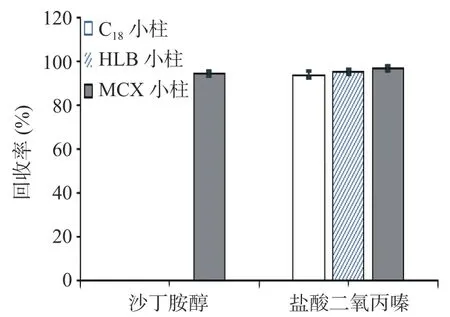
图2 不同固相萃取柱对待测物回收率的影响(n=3)Fig.2 Effectsof different SPE columnson the recoveries of subjects (n=3)
2.3 洗脱溶剂优化
洗脱溶剂的选择是影响待测物质回收率的一个重要因素。本试验选取不同比例的氨水与甲醇洗脱液对回收率的影响进行考察。氨水与甲醇比例分别设置为0.5:100(v/v),1:100(v/v),3:100(v/v),5:100(v/v),10:100(v/v)和100%甲醇。由图3可知,100%甲醇洗脱的加标待测样品中沙丁胺醇回收率为0,盐酸二氧丙嗪回收率为90%(RSD<5%)以上,而0.5%氨水甲醇洗脱的加标待测样品回收率为82.9%~83.6%,RSD为3.1%~4.0%,随着氨水的比例提高,待测样品加标回收率也相应提高,当氨水比例达到3%时,两种待测物质回收率均在90%(RSD<5%)以上。因为沙丁胺醇分子结构含有氨基结构,碱性洗脱剂通过改变离子强度和破坏分子间力,使目标化合物被洗脱下来,得到了较高的回收率。综合考虑,实验最终选择3%氨水甲醇为洗脱溶液,可在不影响待测化合物回收率的前提下,较好地的去除提取液中的极性杂质,净化基质。
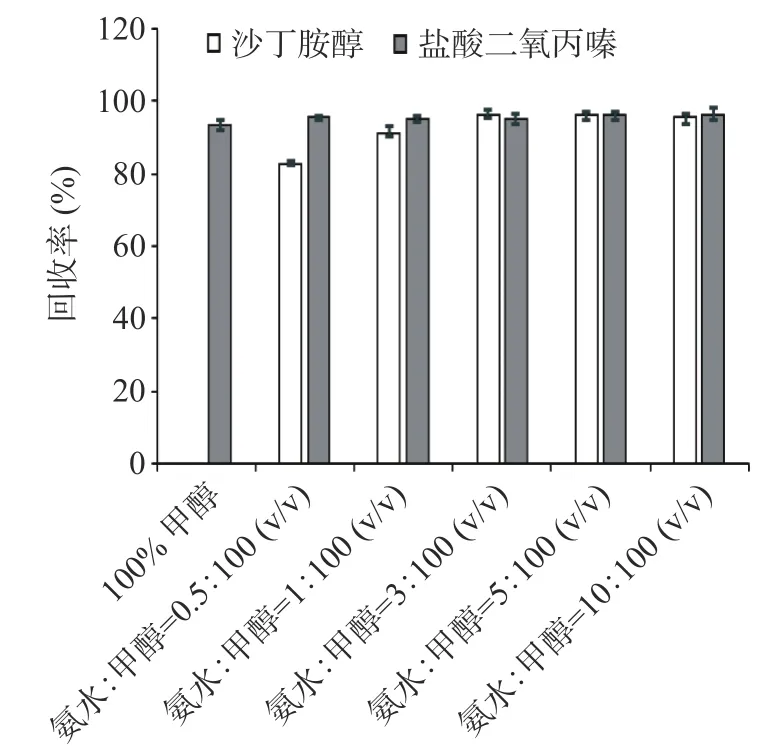
图3 不同比例的氨水和甲醇洗涤液对回收率的影响(n=3)Fig.3 Effectsof different proportionsof ammonia water and methanol on the recovery (n=3)
2.4 色谱柱的选择
本实验考察了ACQUITY UPLC BEH C18(2.1 mm×50 mm,1.7 μm)、ZORBAX SB-C18(2.1 mm×50 mm,1.8μm)以及ACQUITY UPLC BEH HILIC-C18(2.1 mm×50 mm,1.7μm)3种色谱柱的分离效果,结果见图4。在同等色谱条件下,沙丁胺醇和盐酸二氧丙嗪在3种色谱柱上都可以得到较好的分离。但是沙丁胺醇在ACQUITY UPLC BEH C18(2.1 mm×50 mm,1.7μm)柱上峰形对称,相应较高,且保留时间最短。这是因为ACQUITY UPLC BEH C18是端基封尾的色谱柱,且固定相无残留的硅羟基,可以改善峰形,同时耐受p H更广。因此选择ACQUITY UPLC BEH C18(2.1 mm×50 mm,1.7μm)色谱柱进行分离。
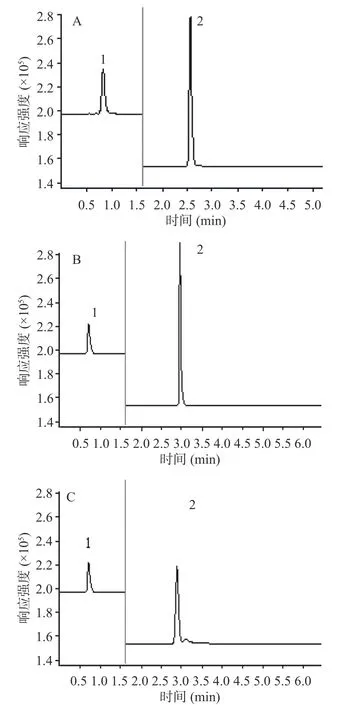
图4 色谱柱分离效果对比Fig.4 Comparison of chromatographic column separation effect
2.5 流动相的选择
流动相是影响色谱峰形状和分离度的一个重要条件。本实验对同一浓度的混合标准曲线溶液,分别考察了甲醇-0.1%甲酸,甲醇-10 mmol/L甲酸铵和甲醇-0.1%甲酸溶液(10 mmol/L甲酸铵)系统。在流动相中加入挥发性有机酸即可以提高离子化效率,同时也有利于样品的溶解,故在流动相中添加甲酸。结果显示,尽管3个系统都可以得到较好的峰形和分离度,但是正离子模式下,在水溶液中添加一定浓度的甲酸能够有效地提高分析物离子化的效率[16],试验以甲醇-0.1%甲酸溶液(10 mmol/L甲酸铵)对待测物进行梯度洗脱,获得的峰灵敏度更高。在保证实验灵敏度的前提下,最终采用甲醇−0.1%甲酸溶液(10 mmol/L甲酸铵)系统。
2.6 子离子选择及碰撞能优化
根据目标物的性质,沙丁胺醇和盐酸二氧丙嗪较易得到一个质子得到母离子,所以2种化合物均采用正离子模式,分别得到一个质子形成母离子[M+H]+。在正离子模式下,分别进行一级质谱扫描(Q1扫描)、二级质谱扫描(子离子扫描)和多反应监测(MRM)扫描。在Q1扫描中找出每个化合物的母离子,在子离子扫描中找到响应较大的子离子,最后在MRM 扫描下优化碰撞能参数,选择响应较高、稳定性好的1个子离子作为定量离子,1个子离子作为定性离子。结果见图5~图7。
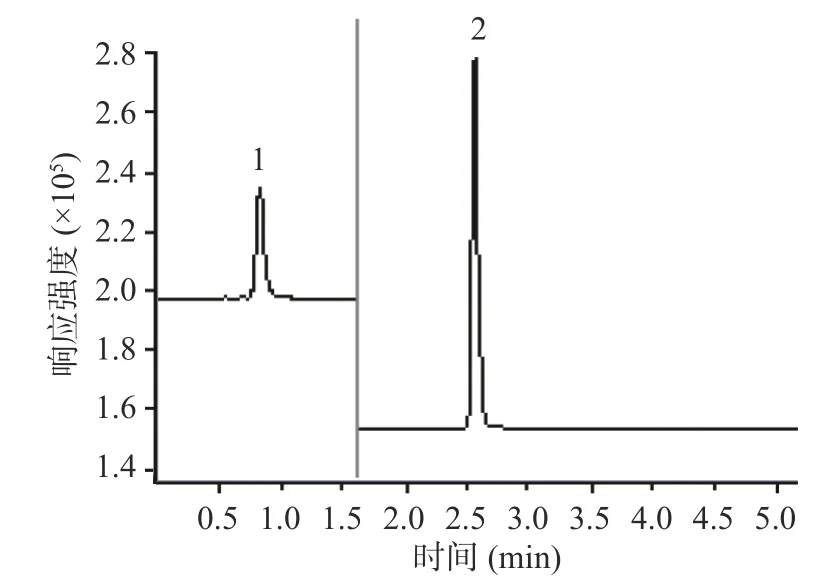
图5 混合对照品溶液PI图Fig.5 Mixed control solution picture of PI
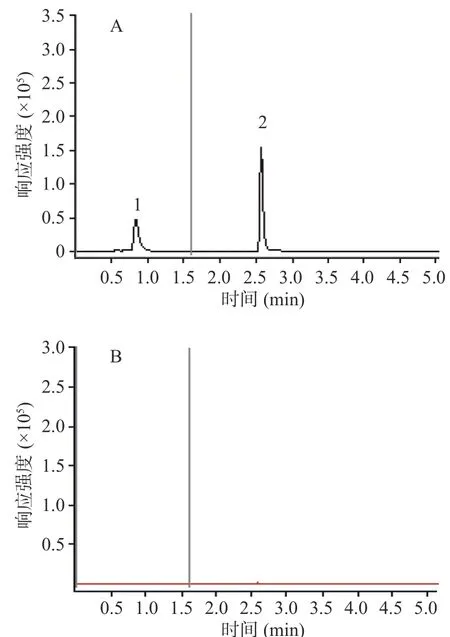
图7 混合标准曲线溶液和供试品(含片Ⅰ)的MRM质谱图Fig.7 Mixed standard curve solution and sample (LozengeⅠ)solution pictures of MRM
2.7 基质效应影响

图6 混合对照品对应二级MS质谱图Fig.6 Mixed control solution PI pictures of secondary mass spectrum
采用质谱进行定量分析时需要考虑基质效应的影响。保健食品基质较为复杂,经简单处理后依然有多种非待测物质保留在样品溶液里[34],进入电离源时会对目标物的离子化过程产生干扰,对检测方法的灵敏度和准确度造成一定影响,进而影响试验的检测结果。本试验采用基质加标标准曲线定量法:添加40 mg/L混合标准溶液制成基质加标液,与流动相加标溶液进行比较,空白基质中标准物质的峰面积(A)和甲醇中标准物质的峰面积(B)的比值考察基质效应(ME),ME=A/B×100%,当ME<1时,说明存在基质抑制作用;当ME>1时,说明存在基质增强作用[16]。实验结果表明,盐酸二氧丙嗪在保健品基质中的ME为0.94~0.98,而盐酸二氧丙嗪在保健品基质中的ME为0.78~0.83,盐酸二氧丙嗪存在一定的抑制效果,为消除基质效应的影响,因此本实验采用空白基质加标标准曲线定量法。
2.8 标准曲线和检测下限
本实验采用阴性空白基质配制混合标准曲线溶液。以各定量离子色谱峰的峰面积为纵坐标,对照品的浓度为横坐标,制作标准曲线。以3倍信噪比(S/N)计算得到检出限(LOD);以10倍信噪比(S/N)计算得到定量限(LOQ)。各回归方程和相关系数见表4。结果显示这2种化学物质在相应的浓度范围内呈良好的线性关系,决定系数R2均大于0.999,表明本方法可以对含有这2个化学成分的样品进行定量分析。

表4 2个化学成分的线性回归方程及检出限和定量限Table 4 Linear regression equations,LOD and LOQof two components
2.9 精密度和重复性实验
取同一浓度混合标准曲线溶液验证精密度,按照“1.2.4”和“1.2.5”项下的液相和质谱的条件进行分析,进行6次平行测试,分别计算2个化学成分峰面积的相对标准偏差,沙丁胺醇和盐酸二氧丙嗪的峰面积RSD均在1.3%~2.7%之间,均<5%,说明该实验方法具有良好的仪器精密度,符合分析测定的要求。选取同一加标样品共6份,按照“1.2.2”项下条件进行前处理得到供试品溶液,按照“1.2.4”和“1.2.5”项下的液相和质谱的条件进行分析,同一批进样,结果表明检出的沙丁胺醇和盐酸二氧丙嗪含量的重复性RSD均在1.1%~1.9%之间,均<5%,表明该方法的重现性良好。
2.10 稳定性实验
取同一浓度混合标准曲线溶液测定稳定性,分别于1.0、4.0、10.0、15.0、20.0、24.0 h注入超高效液相色谱-串联质谱仪进行测定,以沙丁胺醇和盐酸二氧丙嗪的各自定量离子峰面积为指标,测定其RSD。结果表明沙丁胺醇和盐酸二氧丙嗪的稳定性RSD分别为3.1%和4.1%。表明沙丁胺醇和盐酸二氧丙嗪在24 h内基本稳定。
2.11 加标回收实验
精密称取阴性空白样品(片剂、糖果和液体剂型)各9份,按照1倍检出限(2μg/kg)、3倍检出限(5μg/kg)和5倍检出限(10μg/kg)加入适量浓度的混合标准曲线溶液,按照“1.2.2”项下条件制备供试品溶液,按“1.2.4”和“1.2.5”项下的液相和质谱的条件进行分析,测定峰面积,如表5所示,结果得片剂中沙丁胺醇和盐酸二氧丙嗪的回收率范围分别为90.8%~95.2%和92.8%~98.7%,RSD分别为1.2%~2.0%和0.83%~3.1%;糖果剂型中沙丁胺醇和盐酸二氧丙嗪的回收率范围分别为87.4%~94.8%和90.1%~98.8%,RSD分别为1.6%~3.9%和2.0%~3.7%;液体剂型中沙丁胺醇和盐酸二氧丙嗪的回收率范围分别为89.9%~95.2%和92.8%~98.6%,RSD分别为1.6%~2.4%和1.2%~1.8%。结果表明,该方法的准确度满足试验需求。
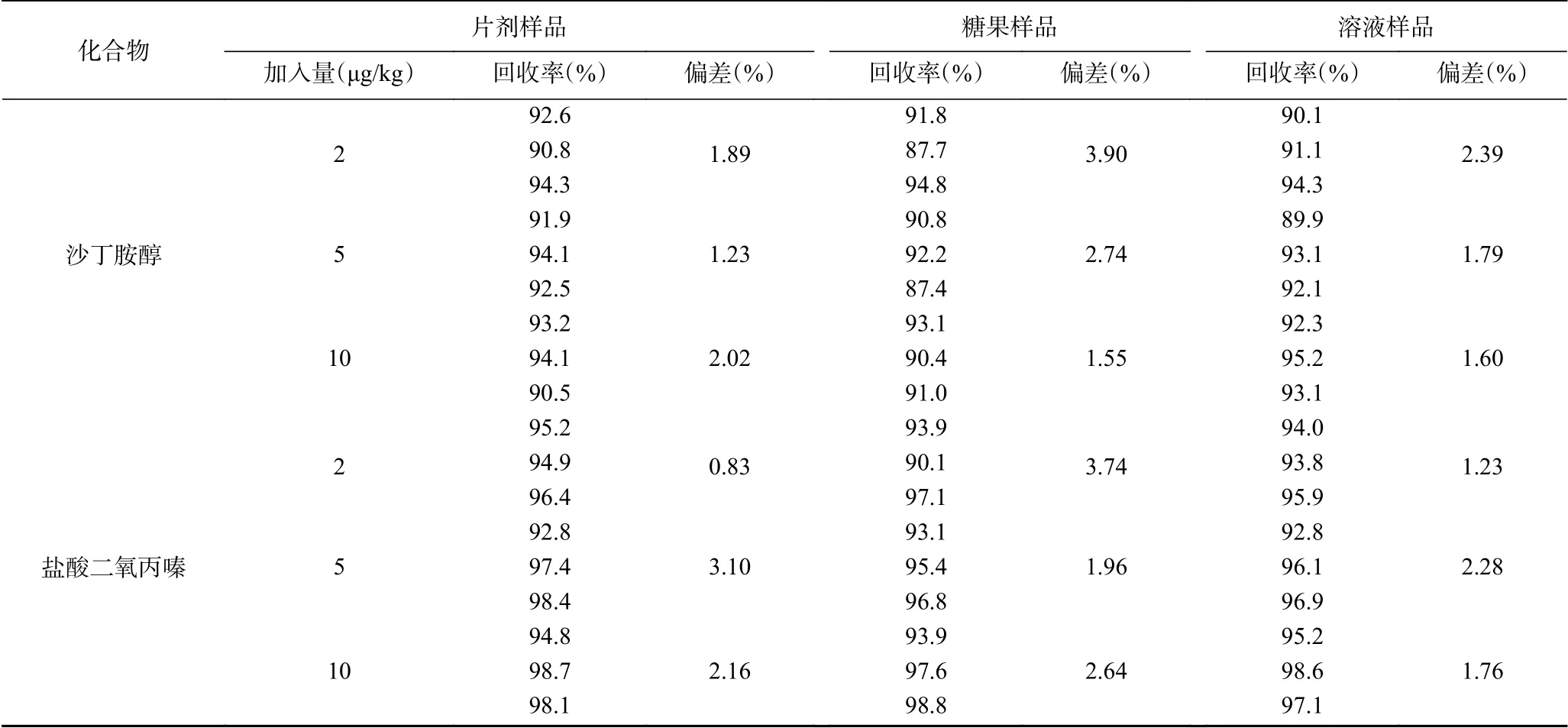
表5 供试品的加标回收率和偏差(n=3)Table 5 Recoveries and RSD(n=3)
2.12 实际样品测定
此次实验选取以含片、糖果以及液体剂型为主的国家抽检和省级抽检保健食品6批次。按照“1.2.2”项下条件备供试品溶液,按“1.2.4和1.2.5中的液相和质谱的条件进行分析检测沙丁胺醇和盐酸二氧丙嗪。含片、糖果以及液体样品均未检出沙丁胺醇和盐酸二氧丙嗪。
3 结论
本实验采用UPLC-MS/MS法,结合全自动固相萃取仪,建立清咽类保健食品中沙丁胺醇和盐酸二氧丙嗪的同时快速测定方法,外标法定量,并对相关条件进行优化。相比于传统人工过固相萃取柱的方法,本试验建立了基于混合型阳离子固相萃取柱的全自动固相萃取的前处理方法,实现自动化,减少人力本,减少人工误差,实现了固相萃取流速的可控性,提高了试验的重复性和准确性,缩短了样品处理周期,适用于大批量样品的前处理。该方法同时对色谱和质谱条件进行了优化,对清咽类保健品中非法添加的沙丁胺醇和盐酸二氧丙嗪进行定性和定量分析。故本文方法的建立能为检测清咽类保健食品中非法添加化学成分检测提供了理论基础,对提高保健食品安全事件的应急处理能力具有重要的参考意义。
猜你喜欢
日用电器(2022年3期)2022-04-14
中国土壤与肥料(2021年5期)2021-12-02
化工自动化及仪表(2021年3期)2021-06-04
食品工程(2020年4期)2021-01-20
今日农业(2020年22期)2020-12-14
食品安全导刊(2020年20期)2020-12-03
食品安全导刊·中旬刊(2020年7期)2020-09-02
中国油脂(2020年3期)2020-04-10
酿酒科技(2019年10期)2019-11-12
中成药(2018年12期)2018-12-29
