
手性1-芳基四氢异喹啉类衍生物的合成新方法
2021-09-01 12:45:58高豪泽王佳欣杨雪柔刘瑞霞聂慧芳
合成化学 2021年8期
高豪泽, 王佳欣, 杨雪柔, 刘瑞霞, 聂慧芳*
(1. 第四军医大学 药学院,陕西 西安 710032; 2. 西安医学院 药学院,陕西 西安 710021)
手性1-芳基四氢异喹啉是一类在天然产物和药物活性分子中广泛分布的药理活性优势骨架,例如cryptostyline II(1)[1]、 cryptostyline III[1](2)、如非竞争性AMPA受体拮抗剂[2](3)、强效TRPM8离子通道受体拮抗剂[3](4)、雌激素受体降解拮抗剂[4](5)和治疗膀胱多动症药物索利那新[5](6)等结构中都包含此类骨架(Chart 1)。因此,手性1-芳基四氢异喹啉骨架的构建一直受到广泛关注,现有合成方法一般是通过关环反应构建异喹啉骨架,如Bischler-Napieralski、 Pictet-Grams和Pomeranz-Fritish法等。环化产物再经过还原反应即可得到四氢异喹啉,或者直接经Pictet-Spengler法合成四氢异喹啉。然而这类方法不但总路线较长且收率低,最关键的是得到的产物均为外消旋体。

Chart 1
2011年,张绪穆等[6]报道了铱催化1-芳基-3,4-二氢异喹啉的不对称氢化,获得最高99%ee的对映选择性。Ratovelomanana-Vidal等[7]于2013年分别以Noyori催化剂、甲酸三乙胺共沸物为手性催化剂和氢源实现了1-芳基-3,4-二氢异喹啉的不对称转移氢化反应,反应获得较好的对映选择性(82%~99%ee)。 2017年,Qu等[8]用亚胺还原酶也实现了这类底物的高对映选择性还原。总的来说,以1-芳基-3,4-二氢异喹啉类化合物为底物制备手性四氢异喹啉衍生物的研究已经获得了许多进展,但是这类亚胺底物的制备要用到一些刺激性大、腐蚀性强的酰氯、三氯氧磷或三氟甲磺酸酐等试剂,不符合环保、可持续发展等绿色合成理念。
近年来,分子内不对称还原胺化反应获得了长足的发展,多个课题组[9-10]应用手性金属催化剂实现了1-芳基四氢异喹啉类化合物的不对称合成,取得了较好的效果。通过脱氨基保护、环化和不对称氢化可以“一锅法”合成1-芳基四氢异喹啉类化合物,这个新策略提供了一种制备该类重要手性砌块的新方法。本文以1,2,3,4-四氢异喹啉为原料,依次与二碳酸二叔丁酯、亚氯酸钠反应得到N-Boc-1,2,3,4-四氢-1-异喹啉酮,再和芳基格氏试剂反应得到分子内不对称还原胺化反应的底物,最后经分子内不对称还原胺化反应可以制备手性1-芳基四氢异喹啉类衍生物(11a~11h, Scheme 1)。

Scheme 1

Scheme 2
1 实验部分
1.1 仪器与试剂
WRS-2型微机熔点仪;Perkin- Elmer 343型自动旋光仪;Bruker 400 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);Agilent 1260型高效液相色谱仪;Daicel CHIRALCEL AS-H型色谱柱。
1,2,3,4-四氢异喹啉、二碳酸二叔丁酯、亚氯酸钠和格氏试剂,萨恩化学技术(上海)有限公司;其余所用试剂均为分析纯。
1.2 合成
(1) 3,4-二氢异喹啉-2(1H)-羧酸叔丁酯(8)的合成[11]
向1,2,3,4-四氢异喹啉(100 mmol, 13.4 g)的二氯甲烷溶液(200 mL)中加入三乙胺(300 mmol, 41.7 mL),滴加二碳酸二叔丁酯(24.0 g, 110 mmol)的二氯甲烷溶液(50 mL),滴毕,反应15 h。减压蒸除溶剂,剩余物经硅胶柱层析[洗脱剂:V(乙酸乙酯)/V(石油醚)=1/10]纯化得化合物823.07 g:白色固体,收率99%, m.p.36.5~38.0 ℃;1H NMR(400 MHz, CDCl3)δ: 7.22~7.04(m, 4H), 4.57(s, 2H), 3.80~3.52(m, 2H), 2.83(t,J=6.1 Hz, 2H), 1.49(s, 9H)。
(2) 1-氧代-3,4-二氢异喹啉-2(1H)-羧酸叔丁酯(9)的合成[12]
向化合物8(50 mmol, 11.65 g)的CH3CCl3-H2O(4/1,V/V)混合溶液(500 mL)中加入NaClO2(150 mmol, 17.0 g),加毕,剧烈搅拌下于55~65 ℃(浴温)反应3 h。冷却至室温,加入饱和NaS2O3溶液(400 mL)淬灭反应,用二氯甲烷萃取,有机层真空干燥,减压蒸除溶剂,剩余物经硅胶柱层析[洗脱剂:V(乙酸乙酯)/V(石油醚)=1/3]纯化得化合物911.5 g:白色固体,收率93%, m.p.71.5~72.9 ℃;1H NMR(400 MHz, CDCl3)δ: 8.30~8.04(m, 1H), 7.52~7.43(m, 1H), 7.40~7.31(m, 1H), 7.21(d,J=7.7 Hz, 1H), 4.09~3.81(m, 2H), 3.01(t,J=6.1 Hz, 2H), 1.59(s, 9H)。
(3) 化合物10a~10h的合成(以10a为例)[9]
于0 ℃惰性气体氛围下,向9(6 mmol, 1.48 g)的THF(20 mL)溶液中缓慢滴加格氏试剂(7.2 mmol),滴毕,搅拌15 min后撤去冰浴,于室温反应过夜。用冰水淬灭反应,乙酸乙酯萃取,有机层真空干燥,减压蒸除溶剂,剩余物经硅胶柱层析[洗脱剂:V(乙酸乙酯)/V(石油醚)=1/5]纯化得化合物10a1.66 g:无色油状液体,收率85%;1H NMR(400 MHz, CDCl3)δ: 7.80(d,J=7.6 Hz, 2H), 7.60(t,J=7.4 Hz, 1H), 7.47(t,J=7.7 Hz, 3H), 7.39(d,J=7.7 Hz, 1H), 7.35~ 7.26(m, 2H), 5.02(s, 1H), 3.39(q,J=6.6 Hz, 2H), 2.86(t,J=6.9 Hz, 2H), 1.40(s, 9H)。
用类似的方法合成10b~10h。
10b[9]: 无色油状液体,收率87%;1H NMR(400 MHz, CDCl3)δ: 7.50~7.34(m, 3H), 7.34~7.23(m, 3H), 7.21(q,J=8.1 Hz, 2H), 5.04(s, 1H), 3.44(q,J=6.8 Hz, 2H), 3.00(t,J=7.2 Hz, 2H), 2.45(s, 3H), 1.41(s, 9H)。
10c[9]: 无色油状液体,收率85%;1H NMR(400 MHz, CDCl3)δ: 7.64(s, 1H), 7.56(d,J=7.8 Hz, 1H), 7.50-7.36(m, 3H), 7.36~7.24(m, 3H), 5.09(s, 1H), 3.41~3.36(m, 2H), 2.91~2.73(m, 2H), 2.39(s, 3H), 1.40(s, 9H)。
10d[9]: 无色油状液体,收率87%;1H NMR(400 MHz, CDCl3)δ: 7.83~7.60(m, 2H), 7.52~7.41(m, 1H), 7.37(d,J=7.9 Hz, 1H), 7.33~7.14(m, 4H), 4.98(s, 1H), 3.37(q,J=7.2 Hz, 6.5 Hz, 2H), 2.83(t,J=7.1 Hz, 2H), 2.43(s, 3H)。
10e[9]: 无色油状液体,收率81%;1H NMR(400 MHz, CDCl3)δ: 7.60~7.35(m, 5H), 7.34 ~7.22(m, 3H), 4.91(s, 1H), 3.38(q,J=6.6 Hz, 2H), 2.86(t,J=7.0 Hz, 2H), 1.40(s, 9H);19F NMR(376 MHz, CDCl3)δ: -111.74。
10f[9]: 无色油状液体,收率83%;1H NMR(400 MHz, CDCl3)δ: 7.84(dd,J=8.4 Hz, 5.5 Hz, 2H), 7.50~7.43(m, 1H), 7.39(d,J=7.7 Hz, 1H), 7.29(d,J=4.3 Hz, 2H), 7.13(t,J=8.4 Hz, 2H), 4.96(s, 1H), 3.37(q,J=6.6 Hz, 2H), 2.84(t,J=7.0 Hz, 2H), 1.40(s, 9H);19F NMR(376 MHz, CDCl3)δ: -104.50。
10g[9]: 无色油状液体,收率81%;1H NMR(400 MHz, CDCl3)δ: 7.79(s, 1H), 7.66(d,J=7.8 Hz, 1H), 7.57(d,J=8.1 Hz, 1H), 7.51~7.45(m, 1H), 7.44~7.36(m, 2H), 7.34~7.27(m, 2H), 4.97(d,J=6.7 Hz, 1H), 3.39(q,J=6.8 Hz, 2H), 2.87(t,J=7.1 Hz, 2H), 1.40(s, 9H)。
10h[9]: 无色油状液体,收率79%;1H NMR(400 MHz, CDCl3)δ: 7.88~7.63(m, 2H), 7.63 ~7.34(m, 4H), 7.29(d,J=4.1 Hz, 2H), 4.98(s, 1H), 3.38(q,J=6.7 Hz, 2H), 2.85(t,J=7.0 Hz, 2H), 1.40(s, 9H)。
(4)11a~11h的合成(以11a为例)[9]
惰性气体氛围下向化合物10(0.5 mmol)的二氯甲烷(3 mL)溶液加入三氟乙酸(0.23 mL, 3 mmol),加毕,搅拌下反应4 h。旋蒸除去易挥发物质,残余物直接用于下步反应。向安瓿瓶中依次加入[Ir(cod)Cl]2(1.6 mg, 2.5 μmol)、tBu-ax-Josiphos(3.7 mg, 5.5 μmol)和THF(1 mL),搅拌30 min得催化剂溶液,向上述溶液中加入40%HBr溶液(7 μL),并将上述脱Boc底物THF溶液(1 mL)加入到催化剂中,最后加入Ti(OiPr)4(0.15 mL, 0.5 mmol),将安瓿瓶移至反应釜,用H2置换3次后升至50 atm,于50 ℃反应20 h。冷却至室温,释放H2,旋干反应液,残余物用EtOAc溶解后用饱和NaHCO3溶液洗涤,真空干燥,减压除去溶剂,剩余物经硅胶柱层析[洗脱剂:V(乙酸乙酯)/V(石油醚)=1/1]纯化得化合物11a98.35 mg:白色固体,收率94%, 95%ee, m.p.95.9~96.9 ℃;1H NMR(400 MHz, CDCl3)δ: 7. 46~7.22(m, 5H), 7.14(d,J=4.4 Hz, 2H), 7.09~6.98(m, 1H), 6.75(d,J=7.8 Hz, 1H), 5.11(s, 1H), 3.39~3.21(m, 1H), 3.17~2.97(m, 2H), 2.92~2.75(m, 1H), 1.98(s, 1H)。
用类似的方法合成11b~11h。
11b[9]: 淡黄色油状物,收率81%, 85%ee;1H NMR(400 MHz, CDCl3)δ: 7.23~7.08(m, 5H), 7.07~6.93(m, 2H), 6.69(d,J=7.7 Hz, 1H), 5.34(s, 1H), 3.42~3.23(m, 1H), 3.21~2.93(m, 2H), 2.90~2.77(m, 1H), 2.41(s, 3H), 1.73(s, 1H)。
11c[9]: 白色固体,收率91%, 89%ee, m.p.85.1~87.2 ℃;1H NMR(400 MHz, CDCl3)δ: 7.21(t,J=7.9 Hz, 1H), 7.18~7.12(m, 2H), 7.12~7.07(m, 2H), 7.07~6.95(m, 2H), 6.76(d,J=7.7 Hz, 1H), 5.06(s, 1H), 3.36~3.21(m, 1H), 3.16~2.95(m, 2H), 2.88~2.73(m, 1H), 2.32(s, 3H), 1.92(s, 1H)。
11d[9]: 白色固体,收率95%, 79%ee, m.p.81.9~83.9 ℃;1H NMR(400 MHz, CDCl3)δ: 7.24~7.07(m, 6H), 7.06~6.99(m, 1H), 6.76(d,J=7.8 Hz, 1H), 5.07(s, 1H), 3.37~3.18(m, 1H), 3.17~2.93(m, 2H), 2.93~2.69(m, 1H), 2.34(s, 3H), 1.73(s, 1H)。
11e[9]: 白色固体,收率94%, 94%ee, m.p.78.4~80.2 ℃;1H NMR(400 MHz, CDCl3)δ: 7.41~7.22(m, 1H), 7.21~7.12(m, 2H), 7.11~7.01(m, 2H), 7.01~6.86(m, 2H), 6.75(d,J=7.7 Hz, 1H), 5.10(s, 1H), 3.31~3.19(m, 1H), 3.14~2.96(m, 2H), 2.89~2.76(m, 1H), 1.90(s, 1H);19F NMR(376 MHz, CDCl3)δ: -113.14。
11f[9]: 白色固体,收率96%, 95%ee, m.p.83.5~85.5 ℃;1H NMR(400 MHz, CDCl3)δ: 7.24(dd,J=8.2 Hz, 5.3 Hz, 2H), 7.15(d,J=4.2 Hz, 2H), 7.09~6.91(m, 3H), 6.72(d,J=7.8 Hz, 1H), 5.09(s, 1H), 3.30~3.20(m, 1H), 3.15~2.98(m, 2H), 2.90~2.73(m, 1H), 1.93(s, 1H);19F NMR(376 MHz, CDCl3)δ: -115.30。
11g[9]: 白色固体,收率92%, 91%ee, m.p.88.9~90.2 ℃;1H NMR(400 MHz, CDCl3)δ: 7.31~7.21(m, 3H), 7.20~7.11(m, 3H), 7.06(dq,J=8.4 Hz, 4.3 Hz, 1H), 6.74(d,J=7.7 Hz, 1H), 5.08(s, 1H), 3.29~3.19(m, 1H), 3.13~2.99(m, 2H), 2.88~2.76(m, 1H), 1.89(s, 1H)。
11h[9]: 白色固体,收率95%, 89%ee, m.p.104.6~106.0 ℃;1H NMR(400 MHz, CDCl3)δ: 7.29(d,J=8.4 Hz, 2H), 7.21(d,J=8.2 Hz, 2H), 7.15(d,J=3.2 Hz, 2H), 7.04(dt,J=8.5 Hz, 4.3 Hz, 1H), 6.71(d,J=7.7 Hz, 1H), 5.08(s, 1H), 3.30~3.20(m, 1H), 3.15~2.98(m, 2H), 2.89~2.75(m, 1H), 1.84(s, 1H)。
2 结果与讨论
2.1 分子内不对称还原胺化反应条件优化
以10a为例,对反应条件进行了优化,结果见表1。由表1可知,Ti(OiPr)4对于反应的转化率和选择性有重要作用,能够将反应的收率和ee值提高至90%和75%ee(Entry 1~2), Ti(OiPr)4可以促进环状亚胺中间体的生成因而可以提高转化率,提高ee值的机理尚不明确。配体筛选结果如Entry 3~6所示,叔丁基取代配体能够获得最高95%ee的对映选择性。酸添加剂对反应的立体选择性有显著影响(Entry 3, 7~8),其中HBr的结果最好。经过反应溶剂筛选可以得出甲苯、二氯甲烷、1,4-二氧六环和乙酸乙酯等非质子性溶剂在10a的分子内不对称还原胺化反应中可以获得较好的收率以及中等至良好的选择性(Entry 9~12)。反应温度和氢气压力对ee值影响性较小,但对转化率影响较大,降低温度或氢气压力都使产物收率下降(Entry13~14)。最终确定了模板反应的最佳反应条件为:以tBu-ax-Josiphos为手性配体,Ti(OiPr)4(1 eq.)和40%HBr溶液(0.1 eq.)为添加剂,四氢呋喃为溶剂,50 ℃反应20 h,催化剂和底物比例为1/100。
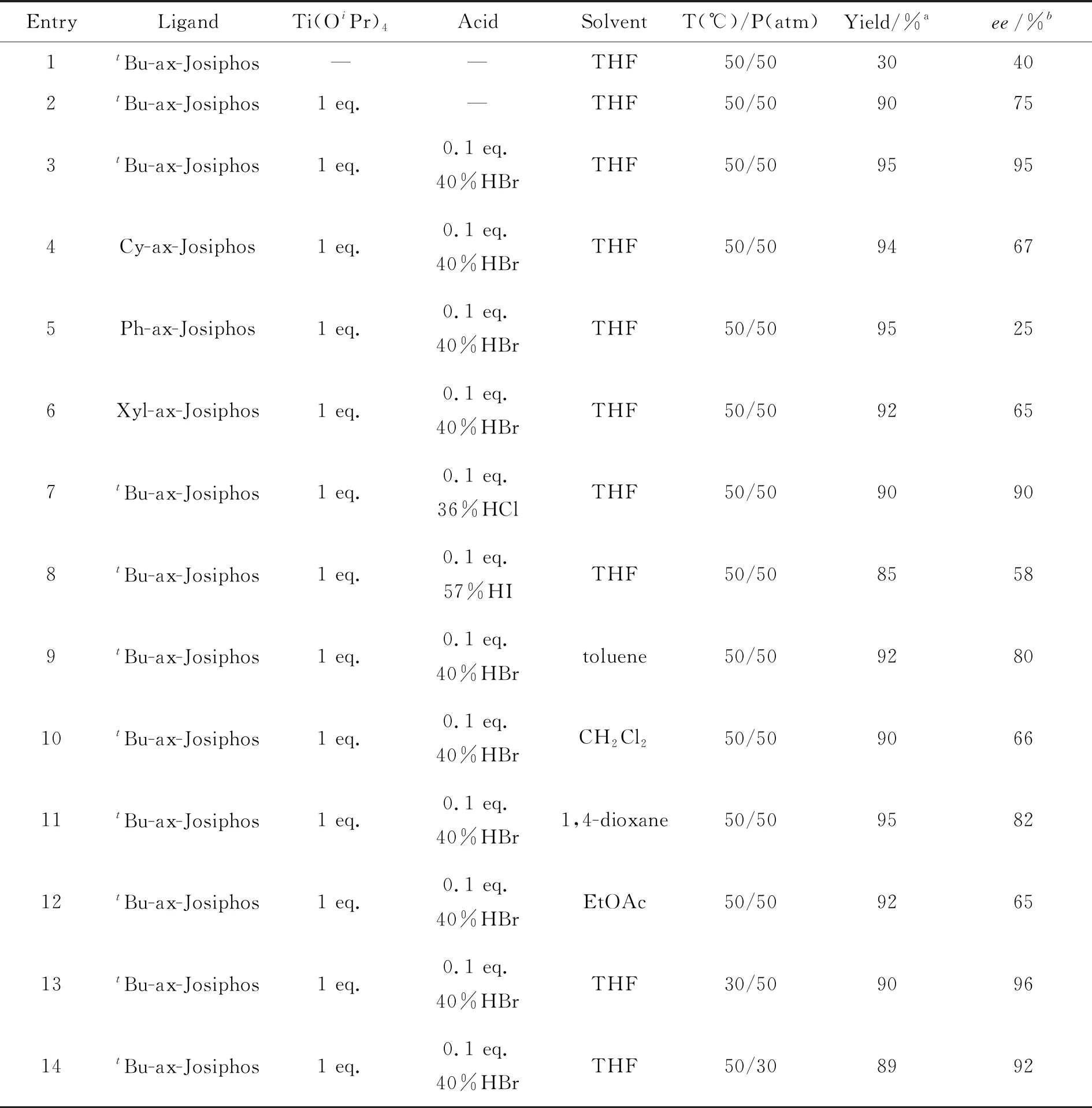
表1 分子内不对称还原胺化反应条件优化
2.2 底物拓展
在最佳条件下,进行底物10的分子内不对称还原胺化反应,结果见Scheme 2。当1-位苯环上取代基位于邻位时,反应收率较低(81%),取代基位于间位和对位时差别较小,都能获得90%以上的收率。当1-位苯环上取代基为吸电性较强的基团时,选择性较高,含F或Cl取代底物结果要好于甲基取代底物。
报道了一种以廉价的1,2,3,4-四氢异喹啉为原料,经分子内不对称还原胺化反应合成重要手性砌块1-芳基四氢异喹啉的新方法。该方法具有原料易得、条件温和且底物适用性较广等优点,为含手性1-芳基四氢异喹啉结构单元的药物和天然产物提供了一种新的合成路径。
猜你喜欢
山西化工(2022年2期)2023-01-14 05:20:05
中成药(2017年7期)2017-11-22 07:33:25
国外医药(抗生素分册)(2016年1期)2016-07-10 12:02:35
合成化学(2015年2期)2016-01-17 09:04:21
合成化学(2015年1期)2016-01-17 08:59:30
化工进展(2015年6期)2015-11-13 00:27:23
化工进展(2015年6期)2015-11-13 00:27:11
中国塑料(2015年10期)2015-10-14 01:13:13
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01 02:54:14
安徽工业大学学报(自然科学版)(2014年4期)2014-07-11 01:45:50
