
人肺癌细胞A549λ噬菌体cDNA文库的构建与鉴定
2021-08-28 09:55张红英郭嘉雯员月明徐志勇邓长生
西南医科大学学报 2021年4期
张红英,郭嘉雯,员月明,徐志勇,邓长生,王 琪
1.广州中医药大学 科技产业园(广州510405);2.广州中医药大学 青蒿研究中心(广州 510405)
肺癌发生于支气管粘膜上皮,亦称支气管癌。近50年来,许多国家都报道肺癌的发病率明显增高[1]。在男性癌瘤病人中,肺癌发病率已居首位;肺癌也占女性常见恶性肿瘤的第2 位或第3 位[2-4]。且目前,肺癌的死亡率仍然居高不下,除了存在被发现晚和诊断晚的因素外,还与没有针对肺癌的特定性基因或靶标蛋白密切相关的药物有关[5-6]。近年来,随着重叠延伸PCR技术(SOEPCR),血清学分析技术(SER⁃EX)和RNA 转录5′末端转换(SMART)技术的兴起,构建肺癌多基因和高质量cDNA表达文库显得非常必要。
mRNA 构建的cDNA 表达文库理论上含有该生物体的所有表达基因,可表达与天然蛋白相似的重组蛋白[7]。因此建立cDNA表达文库在一次性永久保存基因资源的同时,还可以利用功能筛选、免疫学筛选、药物探针筛选、Southern 杂交和大规模序列测定等分子生物学技术寻找特异性活性蛋白,进而通过克隆和表达这些基因,从核酸和蛋白质等分子水平研究药物作用的分子机理,探究药物与靶标的作用机制。
1 材料与方法
1.1 材料
人肺癌细胞A549λ由本实验室提供;TRIzol 试剂为上海碧云天生物技术有限公司产品;SMART™cDNA Library Construction Kit 购 自CLONTECH 公司;λTriplEx2、SfiⅠ、Lambda EcoT14I digest、250 bp DNA Ladder marker 购 自TAKARA 公 司;MaxPlax Lambda Packaging Extracts购自EPICENTRE公司。
1.2 方法
1.2.1 总RNA 提取①人肺癌细胞A549λ生长至90%汇合后,弃培养液,向培养皿中加入3 mL Trizol试剂,轻轻晃动3~5次,移液器吹打几次,确保细胞全部裂解,吸入离心管中;②室温放置5 min,使细胞充分裂解;③加入0.6 mL 氯仿,涡旋混匀,室温放置2~3 min;④12 000 g,4 ℃离心15 min,吸取含总RNA的上层无色水相至一新的离心管;⑤加入1.5 mL 异丙醇,颠倒数次混匀,室温沉淀10 min。12 000 g,4 ℃离心10 min,在管底可见RNA 沉淀,弃上清;⑥加入3 mL 75%乙醇(DEPC 水配制),涡旋混匀,7 500 g,4 ℃离心5 min,弃上清,离心>5 000 rpm,离心1 s),小心吸尽液体;⑦待RNA 凉干后,加入20 μL DEPC水溶解,-70 ℃冻存。取少量测定A260/280,并用琼脂糖凝胶电泳鉴定提取效果。总RNA 用Dnase I 处理,除去杂质DNA,琼脂糖凝胶电泳观察除杂结果。
1.2.2 SMART法反转录合成cDNA 根据SMART™cDNA Library Construction Kit 实验步骤将所提总RNA逆转录成cDNA。
第一链的合成 将3 μL总RNA、1 μL SMART IV Oligonucleotide、1 μL CDS III/3′PCR Primer[5′-AT TCTAGAGGCCGAGGCGGCCGACATG-d(T)30N-1N-3’(N=A,G,C,or T;N-1=A,G,or C)]加入0.5 mL离心管中,涡旋混匀后,离心使液体聚至管底,72 ℃孵 育2 min,冰浴2 min后,依次加入2 μL 5 ×First-Strand Buffer,1 μL DTT(20 mM),1 μL dNTP Mix(10 mM),1 μL SMARTScribe™MMLV Reverse Transcriptase,混匀后短暂离心,并于42 ℃孵育1 h,置于冰上备用。
双链cDNA 合成 将11 μL 第一链cDNA,71 μL去离子水,10 μL 10 × Advantage® 2 PCR Buffer,2 μL dNTP Mix,2 μL l 5′ PCR Primer,2 μL CDS III/3′PCR Primer,2 μL 50×Advantage®2 Polymerase Mix依次加入PCR 管中混匀,离心后,加入2 滴矿物油,置于PCR 仪扩增,反应条件如下:①95 ℃预变性2 min;②95 ℃,30 s;55 ℃30 s;68 ℃,45 s;循环扩增25次;③68 ℃,8 min。PCR扩增后,取5 μL PCR产物用琼脂糖凝胶电泳观察结果。
1.2.3 初级文库的获得
1.2.3.1 将合成的双链cDNA切胶回收除去小片段,琼脂糖凝胶电泳鉴定效果。
1.2.3.2 蛋白酶K 的消化①取2 μL 蛋白酶K、50 μL 双链cDNA 至0.5 mL 离心管中,混合。45 ℃孵育20 min,加50 μL去离子水;②加100 μL酚:氯仿:异戊醇,轻轻倒转混匀,1~2 min;③14 000 rpm 离心5 min,将上层水相转移至新的离心管;④加100 μL氯仿:异戊醇至水相,轻轻混合1~ 2 min,14 000 rpm离心5 min,转移上层水相至新的离心管;⑤加入10 μL 3 M醋酸钠、1.3 μL糖原(20 μg/μL)、260 μL 95%乙醇,立即室温14 000 离心20 min,轻轻弃上清;⑥100 μL 80%乙醇洗涤沉淀,弃乙醇,空气干燥约10 min使残留乙醇挥发,加79 μL去离子水重悬沉淀。
1.2.3.3 SfiⅠ酶切将79 μL cDNA,10 μL 10×SfiⅠBuffer,10 μL SfiⅠEnzyme,1 μL 100×BSA加入离心管中混匀,50 ℃孵育2 h。同时,λTriplEx2 载体SfiⅠ酶切。
1.2.3.4 cDNA 与λTriplEx2 载体连接连接体系如下:1 μL cDNA,1μL λTriplEx2,0.5 μL 10 × Ligation Buffer,0.5 μL ATP(10 mM),0.5 μL T4 DNA Ligase,1.5 μL 去离子水,依次加入离心管中,混匀后离心,并于16 ℃孵育过夜。
1.2.3.5 λ噬菌体体外包装按照MaxPlax Lambda Packaging Extracts(EPICENTRE)说明书进行操作。
溶液准备:噬菌体稀释缓冲液(10 mM Tris-HCl(pH 8.3),100 mM NaCl,10 mM MgCl2),LB液体培养基,LB 固体培养基(含1.5%琼脂),LB 顶层琼脂(含0.7%琼脂)。
大肠杆菌铺板准备:在噬菌体包装反应前1 d,挑取单菌落加入50 mL 补充(10 mM MgSO4)LB 培养基,37 ℃震荡培养过夜。在噬菌体包装反应当天,取5 mL 过夜培养的菌体加到50 mL 补充(10 mM Mg⁃SO4+0.2%麦芽糖)LB液体培养基,37 ℃震荡培养,当OD600达到0.8~1.0 h,将菌体4 ℃保存,最多可保存72 h。噬菌体包装反应步骤参考产品说明书。
1.2.4 初级文库的滴定①挑取单克隆至15 mL LB(含麦芽糖/MgSO4)培养基,37 ℃140 rpm 震荡过夜,使OD600达到2.0。5 000 rpm 离心5 min,弃上清,用7.5 mL 10 mM MgSO4重悬沉淀;②37 ℃预热LB/Mg⁃SO4固体培养基;③用λ稀释缓冲液稀释菌体(稀释范围1∶5-1∶20);④加1 μL 稀释的噬菌体至200 μL XL1~Blue过夜培养物,使噬菌体37 ℃吸附10~15 min;⑤加2 mL 融化的LB/MgSO4顶层琼脂,混匀后立即倒入37 ℃预热的LB 固体培养基;⑥室温冷却10 min,置培养皿于37 ℃6~18 h。
计数噬菌斑,并计算噬菌体滴度。计算公式如下:

1.2.5 初级文库的扩增①从初级文库中挑取单克隆至15 mL LB(含麦芽糖/MgSO4)培养基,37 ℃140 rpm 震荡过夜,使OD600达到2.0。5 000 rpm 离心5 min,弃上清,用7.5 mL 10 mM MgSO4重悬沉淀;②37 ℃预热LB/MgSO4固体培养基。③用4 mL 离心管将500 μL 细菌培养物与稀释的噬菌体混匀(使每平皿含有1×105噬菌斑);37 ℃水浴15 min;每管加4.5 mL融化的LB/MgSO4顶层琼脂;快速混匀,将细菌与噬菌体的混合物倒入LB/MgSO4琼脂培养皿,转动使其分布均匀;④室温放置10 min 使琼脂凝固,将平皿放置培养箱37 ℃培养6~18 h,至噬菌体汇合;⑤每皿加入12 mL 1×λ稀释缓冲液,置于4 ℃过夜;噬菌斑被合并形成扩增的文库;平皿置水平摇床上室温50 rpm摇动1 h;⑥收集λ噬菌体裂解物至无菌烧杯中,充分混匀后均分于50 mL 离心管;每管加10 mL 氯仿,涡旋振荡2 min;5 000 g离心10 min,收集上清至新管,盖紧放置于4 ℃。
扩增文库滴度测定同初级文库。扩增文库4 ℃可保存6个月,长期保存(至少1年)应加入DMSO使其终浓度为7%,置-70 ℃,避免反复冻融。
1.2.6 PCR 扩增检测文库质量随机挑选cDNA 文库中16个单克隆噬菌斑进行PCR鉴定。插入片段检测Primer 序列为:5′ Sequencing Primer:5′-TCCGA⁃GATCTGGACGAGC-3′;3′Sequencing Primer,5′-TA ATACGACTCACTATAGGG-3′。
2 结果
2.1 总RNA提取
紫外分光光度计测定A260/280 为1.92。琼脂糖凝胶电泳结果如图1所示,28S rRNA 和18S rRNA 条带清晰,28S rRNA 的浓度接近18S rRNA 的两倍,表明总RNA 无明显降解,效果比较理想。利用Image J软件计算28S和18S灰度值,P<0.05。
总RNA 用Dnase I 处理后,电泳结果如图2 所示,与图1 相比,条带分界明显,表明残留DNA 已被除去,总RNA得以纯化。

图1 总RNA琼脂糖凝胶电泳及其值的比较
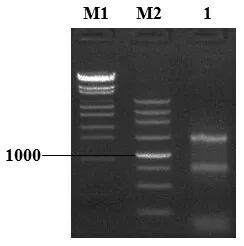
图2 Dnase I纯化总RNA
2.2 双链cDNA的合成
cDNA文库质量评价有两个重要的指标,即文库的库容和重组序列的完整性。初级文库的库容大于有效文库的要求1×106pfu/mL,插入片段分布在500~3 000 bp 之间,大小不同且大片段居多,说明构建的文库完全满足高质量文库的要求。SMART法反转录合成双链cDNA,结果如图3所示,双链cDNA呈弥散分布,条带正常且量足,符合建库需求。
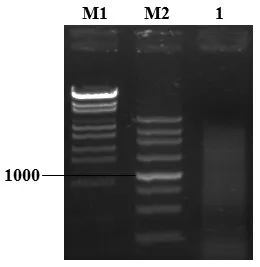
图3 双链cDNA琼脂糖凝胶电泳
双链cDNA 切胶回收除去小片段,琼脂糖凝胶电泳鉴定结果如图4 所示,与图3 比较可见,小于500 bp的小片段已经除去,剩余双链cDNA片段大小位于500~3 000 bp之间。
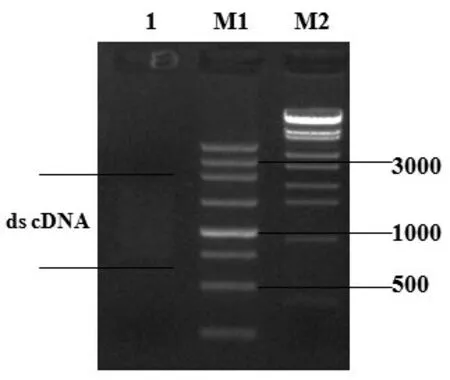
图4 除去小片段的双链cDNA
2.3 文库质量检测
实验测定初级文库的库容为1.88×107pfu/mL,扩增文库滴度为2.50×109pfu/mL。初级文库包装产物约700 μL,扩增噬菌体文库约150 mL。随机挑选cDNA文库中16个单克隆噬菌斑进行PCR鉴定,结果如图5 所示,扩增的片段主要集中在500~3 000 bp之间,表明本实验所构建的cDNA文库质量合格。

图5 cDNA文库随机克隆的PCR产物
3 讨论
cDNA 文库是指生物发生发展过程中某一特定组织或细胞所有基因包含其中的集合体[8],包括标准化cDNA 文库,消减cDNA 文库及RACE cDNA 文库等[9-10],为现代基因组学研究提供了基础。目前,cD⁃NA 文库应用于反向遗传学在分子水平上研究特定阶段不同基因的表达差异,寻求不同发育阶段关键基因,克隆新型细胞因子和分离特异性基因[11-12]。
尽管血清学分析(serological analysis of antigens by recombinant expression cloning,SEREX)是寻找抗原的新方法[13],通过该方法可以克隆多种类型肿瘤基因,食管鳞状细胞癌肿瘤抗原NY-ESO-1[14-15],多发性骨髓瘤相关基因MMSA-1~9[16],MMSA-11~13[16-17],鼻咽癌相关抗原GX-NPC-1~14[18-19]和胃癌肿瘤相关抗原MPS-1[20-21]等。但该技术的局限性在于筛选得到的肿瘤抗原有可能是假阳性,需大量对比检测正常人以及确诊肿瘤病人的血清,结合CD4+和CD8+T细胞多次评估[22-23]。
因此,本实验采用SMART技术成功构建人肺癌细胞A549 λ噬菌体cDNA 文库,具有起始RNA 用量少、cDNA文库重组率高、所含全长cDNA比例高、装载容量大、质量高、适合长期保存等优点。同时,可通过常规电泳对每一步实时监测,避免了传统cDNA文库代表性差、筛选工作量大等弊端。经鉴定,本实验构建的初级文库库容为1.88×107pfu/mL,扩增文库滴度为2.50×109pfu/mL,插入cDNA片段为500~3 000 bp,符合质量标准。人肺癌细胞A549 λ噬菌体cDNA文库的成功构建,为下一步筛选治疗肺癌的靶标药物奠定了基础。
4 结论
本研究成功构建人肺癌细胞A549 λ噬菌体cD⁃NA 文库,旨在筛选克隆与肺癌相关的相互作用蛋白,深入研究肺癌抗原基因功能,为肺癌的早期诊断和治疗提供理论基础。
猜你喜欢
农产品加工(2022年17期)2022-10-17
化工进展(2022年7期)2022-08-01
昆明医科大学学报(2022年2期)2022-03-29
植物保护(2021年4期)2021-11-12
新农业(2021年9期)2021-06-20
科技视界(2020年26期)2020-09-24
科技视界(2020年17期)2020-07-30
科学24小时(2020年4期)2020-05-14
家禽科学(2020年3期)2020-05-13
新农业(2019年5期)2019-06-28
