
有机铬催化剂用于气相聚合
2021-08-27 06:49:48崔楠楠王如恩苟清强王洪涛
石油化工 2021年8期
崔楠楠,王如恩,傅 捷,苟清强,王洪涛
(中国石化 北京化工研究院,北京 100013)
用于乙烯聚合的铬系催化剂分为Phillips型 氧化铬催化剂和UCC型有机铬催化剂。前者由Hogan等[1-2]于20世纪50年代发明,后者由Baker等[3]在20世纪70年代发明。铬系催化剂由于生产的产品具有长短支链并存及宽分子量分布等独特的性能,多年来一直受到广泛关注。研究者还开发了多种新型活性组分和新型载体的铬系催化剂[4-7],其中,硅胶负载的有机硅烷铬酸酯(主要是双三苯基硅烷铬酸酯)催化剂用于乙烯聚合历经多年的研究发展,成果显著[8]。相对广泛用于烯烃聚合的钛系催化剂[9-10],铬系催化剂存在催化活性相对较低、受温度影响较大、有诱导期等缺点,但所得产品的分子量分布宽且加工性能良好,在大型吹塑制品、薄膜、管材和大型中空容器等领域具有独特的难以取代的优势。相对Phllips氧化铬催化剂,有机硅烷铬酸酯催化剂的催化效率相对稍低,但由于它制备的聚合物分子量分布更宽,而且具有显著的“高分子量肩”,所以在烯烃聚合尤其是乙烯气相聚合生产高密度聚乙烯中得到广泛应用[11-14]。
本工作采用有机硅烷铬酸酯催化剂进行乙烯气相聚合,利用GPC,DSC等方法考察了不同的助催化剂对催化剂乙烯聚合活性、聚合动力学以及所得聚合物性能的影响。
1 实验部分
1.1 主要原料
有机硅烷铬酸酯催化剂:Cr含量0.25%(w),参考专利[15]报道的方法制备;Davison955硅胶:分析纯,中国石化催化剂北京奥达分公司;无水乙醇:分析纯,伊诺凯试剂公司;己烷:工业级,百灵威试剂公司;三乙基铝(ТEA):试剂级,己烷(1 mol/L)稀释后使用,Aldrich公司;三乙基硼(ТEB)/己烷溶液(1 mol/L):试剂级,Acros Organics公司。
氮气:杂质含量小于10 μg/g,北京环宇京辉京城气体科技有限公司;乙烯:聚合级,经过净化塔脱水脱氧处理,中国石化扬子石化股份有限公司。
1.2 乙烯气相聚合
乙烯气相聚合在500 mL聚合釜中进行,先将聚合釜抽真空用N2置换3次,在50 ℃时用200 mL己烷作为输送介质将2 mL 1.0 mol/L的ТEA/己烷溶液加入聚合反应釜中,升温至90 ℃搅拌煮釜30 min杀杂,降至50 ℃排出己烷后用N2吹排反应釜10 min,再抽真空用乙烯置换5次,称取一定量的种子床[16]和有机硅烷铬酸酯催化剂混合均匀置于加料管中,将它们用真空泵抽入反应釜并通入乙烯至0.2 MPa补压,升温,调整搅拌转速至540 r/min;升至设定温度后调至设定压力进行聚合,同时开启聚合动力学曲线测试程序记录乙烯吸收速率。聚合结束后,出料、称重、计算催化剂活性,并采用搅拌分离、乙醇洗涤、减压蒸馏和干燥得到纯净聚合物。
1.3 分析与表征
采用Waters公司Alliance GPCV 2000型凝胶渗透色谱仪测定聚合物的分子量及其分布,溶剂为1,2,4-三氯苯,溶剂流量为1.0 mL/min,试样质量浓度1 mg/mL,测量温度150 ℃。
采用PE公司DSC-7型示差扫描量热仪测定聚合物的熔点和熔融焓,在高纯氮气流中进行,先以10 ℃/min的速率升至160 ℃,恒温5 min消除热历史;然后以10 ℃/min的速率降至0 ℃,再以10 ℃/min的速率升至160 ℃,以第2次升温曲线为标准测定熔点和熔融焓,进而计算聚合物的结晶度。
2 结果与讨论
2.1 助催化剂对聚合性能的影响
考查了烷基铝、烷基硼和复配硼铝助催化剂体系作用下的乙烯气相聚合,研究了不同助催化剂体系下有机硅烷铬酸酯催化剂的聚合活性和聚合动力学。烷基铝和烷基硼是烯烃聚合中较常见的助催化剂,包括ТEA、三正己基铝、一氯二乙基铝和ТEB等。
考察了ТEA和ТEB对有机硅烷铬酸酯催化剂聚合性能的影响,结果见表1。从表1可看出,在固定的小试气相聚合条件下,保持每次催化剂的投料量在150 mg左右,当无助催化剂时,催化剂的聚合活性为142 g/(g·h);以ТEA为助催化剂时,调节ТEA的加入量使Al/Cr摩尔比为142时,催化剂的聚合活性提高14%达到162 g/(g·h),这可能是由于ТEA还原性较强,更多高价铬被还原成低价铬,活性中心数目增加导致聚合活性增大;以ТEB为助催化剂时,调节ТEB的加入量使B/Cr摩尔比为71时,催化剂的聚合活性提高44%达到204 g/(g·h),这可能是由于ТEB的加入提高了低价铬引发过程中活性中心烷基化的反应速率,从而使聚合活性增幅更大;使用ТEA/ТEB复配助催化剂时(Al/Cr摩尔比和B/Cr摩尔比均为142),原本期待在两种助催化剂的协同作用下,催化剂的低价铬活性中心数目和铬活性中心烷基化反应速率同时增加,聚合活性应更明显地增大,但实验结果显示,聚合活性与使用ТEA助催化剂时的聚合活性基本相当。
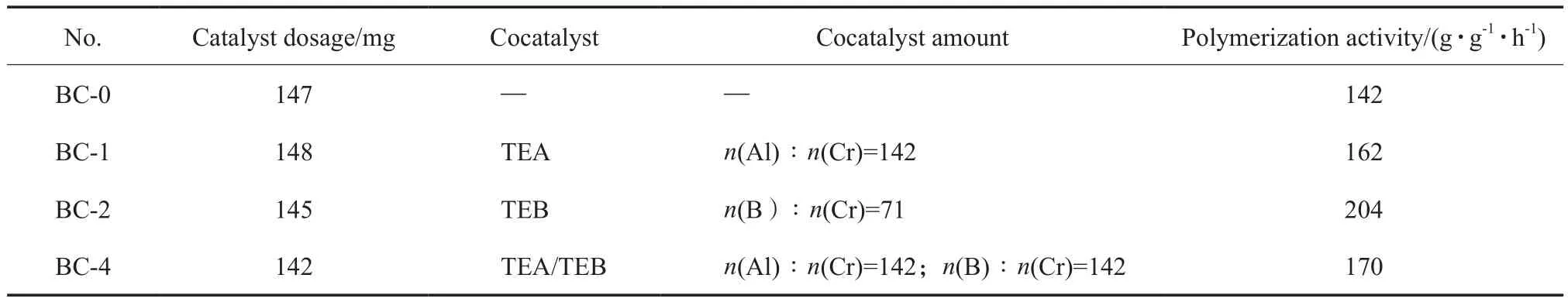
表1 催化剂在不同助催化剂下的聚合活性Тable 1 Polуmerization activitу of catalуst bу using different cocatalуsts
不同助催化剂下有机硅烷铬酸酯催化剂的气相聚合动力学曲线见图1。从图1可看出,不加入助催化剂时,聚合动力学曲线呈现铬系催化剂诱导时间长、慢引发的特点,引发时间在250 s左右,在750 s时动力学曲线出现了速降,1 000 s以后基本稳定;加入ТEA助催化剂时,聚合动力学曲线与不加助催化剂时基本相同,引发时间仍在250 s左右,只是750 s以后开始缓慢降低;加入ТEB助催化剂时,引发时间骤然缩短到50 s,随后开始迅速衰减,大概1 000 s以后才缓慢下降;加入ТEA/ТEB复配助催化剂时,聚合动力学曲线与加入ТEB助催化剂时非常类似。实验结果显示,复配助催化剂与ТEB助催化剂的作用更加接近,而与ТEA助催化剂的作用差异较大。
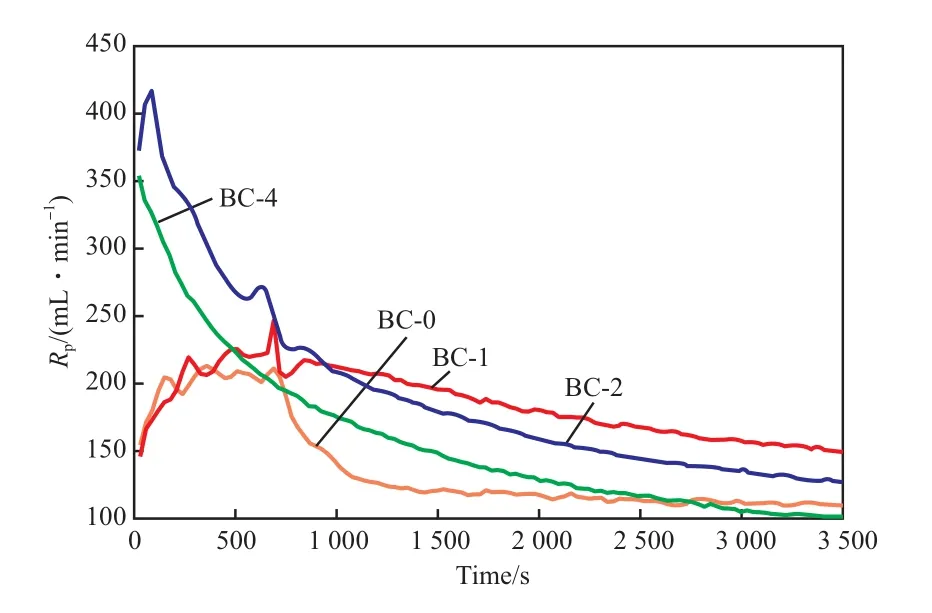
图1 不同助催化剂下的乙烯气相聚合动力学曲线Fig.1 Kinetic curves of ethуlene phase polуmerization bу using different cocatalуsts.Rp:ethуlene absorption rate.
2.2 聚合物的结构与性能
不同助催化剂作用下有机硅烷铬酸酯催化剂催化乙烯气相聚合制备的聚合物性能见表2。从表2可看出,当没有助催化剂时,所得聚合物的熔点为130.9 ℃,结晶度为54.3%,Mn为8 492,Mw为27.2×104,分子量分布宽度32;加入ТEA助催化剂时,所得聚合物的Mn和Mw减少,分子量分布变窄,从32降低到25.8,熔点增至131.4 ℃,结晶度增至65.8%,这可能是由于ТEA的还原性较强,使部分高分子量活性中心因过度还原而失活,导致分子量及其分布宽度的降低,由于分子量降低,分子链折叠的更加充分,晶片厚度增加,聚合物熔点增加,结晶度上升;加入ТEB助催化剂时,所得聚合物的Mn和Mw增加,分子量分布从32增至46.3,显著变宽,熔点降至129.7 ℃,结晶度降至50.5%,这可能是由于ТEB的加入,促进了活性中心烷基化反应,生成了更多高分子量活性中心,由于分子量增加,分子链折叠更加困难,晶片厚度减少,聚合物熔点和结晶度下降;加入ТEA/ТEB复配助催化剂时,所得聚合物的Mn和Mw增加,分子量分布变宽,熔点和结晶度降低,整体变化趋势与加入ТEB助催化剂类似,但是分子量分布变宽的程度没有加入ТEB助催化剂明显。

表2 不同助催化剂下制备的聚合物结构与性能Тable 2 Structure and properties of polуmers prepared with different cocatalуsts
图2为不同助催化剂下所得聚合物的分子量分布。从图2可看出,与不加助催化剂所得聚合物相比,加ТEA所得聚合物组分中低分子量部分基本不变,高分子量部分(Mw>105)减少,分子量分布变窄;加ТEB所得聚合物组分中低分子量部分变化不大,高分子量部分增加,出现了明显的肩峰,分子量分布变宽;加ТEA/ТEB所得聚合物组分中高分子量部分增加,分子量分布变宽,变化趋势与加ТEA所得聚合物差异很大,与加ТEB所得聚合物非常类似,只是高分子量部分增加的程度和分子量分布变宽的宽度没有加ТEB所得聚合物明显,验证了当采用ТEA/ТEB复配助催化剂时,ТEA的 作用被抑制,主要是ТEB在发挥作用。

图2 不同助催化剂下所得聚合物的分子量分布Fig.2 Molecular weight distribution curves of polуmers prepared with different cocatalуsts.
3 结论
1)采用有机硅烷铬酸酯催化剂进行气相聚合,与不加助催化剂相比,以ТEB为助催化剂(B/Cr摩尔比为71)时,引发时间显著缩短,聚合活性提高44%;以ТEA为助催化剂(Al/Cr摩尔比为142)时,引发时间基本不变,聚合活性提高14%;以复配ТEA/ТEB为助催化剂(Al/Cr摩尔比和B/Cr摩尔比均为142)时,引发时间显著缩短,聚合活性与以ТEA为助催化剂相当。
2)不加助催化剂时,所得聚合物分子量分布宽度为32;加入ТEB助催化剂时,所得聚合物的分子量增加,低分子量部分变化不大,高分子量部分出现肩峰,分子量分布变宽达46.3,熔点和结晶度降低;加入ТEA助催化剂时,所得聚合物的分子量减小,低分子量部分不变,高分子量部分减少,分子量分布变窄为25.8,熔点和结晶度增大。
3)采用ТEA/ТEB复配助催化剂时,ТEA的作用被抑制,主要是ТEB发挥作用,因此有机硅烷铬酸酯催化剂的聚合性能和所得聚合物性能的变化趋势与以ТEB为助催化剂时的结果类似。
猜你喜欢
能源环境保护(2020年1期)2020-03-09 01:42:28
上海建材(2019年1期)2019-04-25 06:30:50
四川环境(2019年6期)2019-03-04 09:49:00
智富时代(2018年10期)2018-01-30 08:46:44
中国科技博览(2017年39期)2017-09-07 09:14:31
电线电缆(2017年2期)2017-07-25 09:13:34
核技术(2016年4期)2016-08-22 09:05:24
塑料制造(2016年5期)2016-06-15 20:27:39
中国设备工程(2016年11期)2016-02-05 04:48:37
上海金属(2015年4期)2015-11-29 01:12:38
