
JAK/STAT3介导肿瘤恶病质肌肉萎缩及药物干预研究进展
2021-08-21 07:56:22桑亚洲陈玉龙吴耀松
中国比较医学杂志 2021年7期
桑亚洲,张 艳,陈玉龙,吴耀松
(河南中医药大学中医药科学研究院 河南省中医方证信号传导重点实验室/河南省中医方证信号
传导国际联合重点实验室,郑州 450046)
肿瘤恶病质是一种以体重减轻和肌肉萎缩为主要特征,伴随或不伴随脂肪减少,并且营养支持不能完全逆转,生理机能减退和功能出现障碍为主要特征的综合征[1],这与机体代谢紊乱密切相关,至今尚无有效的药物干预措施可完全逆转恶病质状态[2-4]。恶病质的公认诊断标准以体重和体重指数(body mass index,BMI)为主要观察指标,即6个月内肿瘤患者非意愿性体重降低>5%;或BMI<20 kg/m2伴体重降低>2%:或者四肢骨骼 肌质量指数(appendicular skeletal muscle index,ASMI)小于7.26 kg/m2(男)或小于5.45 kg/m2(女)伴体重降低>2%[5]。其病理生理学特征是食物摄入不足和代谢异常(包括能量消耗,过度的分解代谢和炎症)引起的机体功能和器官严重衰退。大量文献表明,多个器官参与晚期癌症患者的骨骼肌丢失,如中枢神经系统调节[6]、白色脂肪组织损失[7]、胰腺功能障碍[8]、肝代谢失衡[9]、骨骼和肌肉的相互作用[10]、肠道菌群变化[11]均可以诱发机体能量失衡和体重丢失,导致严重的肿瘤恶病质肌肉萎缩,已成为癌症相关恶病质发展的重要因素[12-13]。JAK/STAT3信号通路是一条多种细胞因子共用的信号传导途径,在调控细胞增殖、凋亡、血管新生等多个方面承担着关键作用,在肿瘤恶病质的发生发展中也发挥重要的调节作用,相关研究表明JAK/STAT3信号通路是引起肿瘤恶病病质肌肉萎缩的关键环节,现总结如下:
1 JAK/STAT3信号通路介导的肿瘤恶病质肌肉萎缩机制
Janus激酶(Janus kinase,JAK),是一个细胞内非受体酪氨酸激酶家族,可介导细胞因子产生的信号,并通过JAK/信号传导及转录激活因子(signal transducers and activators of transcription,STAT)信号通路传递。JAK/STAT信号通路参与将信息从细胞外多肽信号传递到细胞核中的靶基因启动子。该途径是通过细胞因子配体与细胞表面细胞因子受体的结合而启动,配体的结合触发JAK的活化,而活化的JAK可以磷酸化受体上的关键酪氨酸残基,从而通过其SH2结构域(src homology 2,SH2)募集特定的STAT形成二聚体,并在输入蛋白α(importin-α)的转运下进入细胞核与特定的DNA靶标结合进而发挥其调控细胞生长、分化、增殖,凋亡等一系列生理病理作用[14-15]。STAT有多个蛋白家族,包括(STAT1、STAT2、STAT3、STAT5A、STAT5B、STAT6),其中STAT3的蛋白和基因表达是造成肿瘤恶病质肌肉萎缩的关键。肿瘤恶病质相关的临床实验观察中发现一些炎症因子可以活化JAK/STAT3信号通路引起STAT3过表达,相反的,通过使用JAK或STAT3抑制剂可有效减缓肌肉萎缩进程[16]。在C26结肠癌肿瘤恶病质动物模型的骨骼肌中可发现STAT3磷酸化和其靶向核基因被激活,通过激活C2C12成肌细胞中的STAT3表达可诱导肌纤维萎缩并加剧肌肉消耗[17]。
骨骼肌是高度可塑性的组织,当有外源性或内源性刺激时,其质量和代谢功能会发生改变,这种改变是骨骼肌维持正常动态平衡的关键。骨骼肌的动态平衡主要通过分解代谢和合成代谢实现的。合成代谢平衡在生长因子,营养物质利用和身体活动等作用下调节,主要通过胰岛素样生长因子-1(insulin-like growth factors-1,IGF1)/磷脂酰肌醇-3激酶(phosphatidylinositol 3-kinase,PI3K)/蛋白激酶B(protein kinase B,AKT)/雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)信号传导途径和肌生长抑制素(myostatin)-smad2/3途径的激活来促进蛋白质的合成;分解代谢平衡主要通过泛素蛋白酶体途径(ubiquitin-proteasome system,UPS)和自噬体溶酶体途径的激活来促进蛋白质降解导致骨骼肌质量损失[18-19],在肿瘤恶病质的相关研究中,JAK/STAT3信号通路对泛素蛋白酶体途径和自噬途径均有调控作用,可通过降解机体内蛋白质造成骨骼肌大量丢失,是造成肿瘤恶病质肌肉萎缩的两种关键途径。
1.1 JAK/STAT3激活可通过泛素蛋白酶体系统降解蛋白质加速骨骼肌萎缩
UPS是细胞内蛋白质降解的主要途径,参与细胞内80%以上蛋白质的降解。泛素化步骤主要为E1活化泛素分子,活化的泛素分子转移至E2,在E3催化下活化的泛素羧基和靶蛋白赖氨酸侧链的氨基形成异肽键,靶蛋白被泛素化、蛋白酶体识别并降解靶蛋白。其中,E3催化泛素分子转移到靶蛋白上是UPS的限速步骤,而肌肉萎缩基因(muscle atrophy f-box,MAFbx/Atrogin-1)和肌肉特异性环指基因1(muscle ring finger 1,MuRF1)正是导致肌肉降解的E3酶[20]。在C26结肠癌与C2C12成肌细胞共培养中,C2C12肌管中的STAT3(p-STAT3)被激活,且p-STAT3可诱导两种蛋白水解途径,一种是通过CAAT区/增强子结合蛋白δ(CAAT/enhancer-binding proteinδ,C/EBPδ)激活半胱氨酸天冬氨酸蛋白酶3(cysteinyl aspartate specific proteinase,caspase-3),被激活的caspase-3不仅裂解肌动球蛋白,为UPS降解提供底物,还刺激26 S蛋白酶体(UPS中关键蛋白酶)的蛋白质降解,即与UPS相互作用以增加肌肉蛋白质的降解[21];另一种是p-STAT3对UPS(MAFbx/Atrogin-1和MuRF-1)的激活,在体内外均发现,当STAT3被磷酸化后激活刺激MuRF-1和MAFbx/Atrogin-1的基因和蛋白表达增加,而抑制STAT3激活可有效减少蛋白水解,增加肿瘤恶病质小鼠的体重、肌肉质量、肌肉抓力,降低比目鱼肌和趾长肌肌肉中蛋白质的降解率[22]。另外,一项针对C26小鼠腓肠肌萎缩和C2C12成肌细胞的实验发现,通过抑制STAT3的活性可有效减缓C26小鼠的肌肉消耗以及IL-6诱导的C2C12肌管萎缩,其机制可能是当STAT3被抑制后可降低MAFbx/Atrogin-1和MuRF-1的表达,进而有效保持肌肉质量[23]。以上研究表明在肌肉蛋白质降解过程中STAT3介导的UPS发挥关键的作用。
1.2 JAK/STAT3可通过激活自噬溶酶体系统调控自噬并引起肌肉萎缩
自噬的发生是自噬体与溶酶体的融合形成自噬溶酶体,其作用是靶向清除蛋白质和受损细胞器等[24]。自噬主要有四个步骤:包括诱导阶段、启动阶段、延长阶段、成熟降解阶段,主要参与的分子有Atg1/ULK1、Atg6/Beclin1、Atg8/LC3B、p62/SQSTM1。
生理状态下,自噬以一种低速率保持稳态,以维持细胞正常的代谢需要和细胞器的更新,但在不同的应激刺激下,自噬对肌肉质量的保持是双向性的,当自噬被长期抑制时,可引起肌无力、肌萎缩;当自噬过度激活可引起骨骼肌蛋白的水解加速,进而引起肌肉萎缩[25-26]。
STAT3对自噬的调控也是双向性的,胞质中的STAT3单体在Tyr705上被Src或JAK激酶磷酸化,然后形成STAT3二聚体,入核与特定的DNA结合,通过转录激活或抑制自噬相关靶基因,例如B细胞淋巴瘤-2(B-cell lymphoma-2,BCL2)、自噬基因BECN1、低氧诱导因子-1α(hypoxia inducible factor-1α,HIF-1α)和BCL2和腺病毒E1B19×103相互作用蛋白3(bcl2/adenovirus e1b interacting protein 3,BNIP3)基因等,并根据不同的环境刺激抑制或促进自噬。另外,胞质中未磷酸化的STAT3可有效抑制真核翻译始动因子2-α激酶2(eukaryotic translation initiation factor 2-αkinase 2,EIF2αK2)、叉头状转录因子O1(forkhead transcription factor O1,FoxO1)和叉头状转录因子O3(forkhead transcription factor O3,FoxO3)基因,其中EIF2AK基因可通过磷酸化EIF2A来促进自噬;FOXO1和FOXO3基因可通过转录激活一系列自噬相关基因来促进自噬[27]。在三种不同的癌症恶病质实验模型中,通过评估自噬降解途径中具有代表性的标志物BECLIN-1、LC3B、p62/SQSTM1的蛋白和基因表达水平,表明了肌肉萎缩与自噬激活的正相关[28]。通过分析食管癌恶病质患者肌肉中相关自噬基因LC3B、p62/SQSTM1的过表达,说明自噬溶酶体可能通过激活蛋白水解系统参与人类癌症恶病质肌肉萎缩的发展[29]。另外,在肿瘤的发生发展进程中自噬与凋亡也发生相互拮抗作用,自噬通过为肿瘤细胞提供能量,抑制其凋亡进程[30]。
1.3 JAK/STAT3与microRNA-17的相互作用在肿瘤恶病质肌肉萎缩中的机制研究
MicroRNA(miRNA)是细胞基因表达调节剂的一部分,可以显著改变细胞的表型特征,相关的动物研究通过筛选多种microRNA的功能及其在小鼠乳腺癌转移中发挥的作用,表明microRNA在癌症转移中具有相关性[31]。相关miRNA和共调节因子可作为特定靶标,激活JAK/STAT3诱导特定表型表达[32],在一项基于癌症恶病质肌肉消瘦研究的miRNA调控网络Meta分析表明miR-17与肌肉萎缩有关,其机制是miR-17可通过抑制STAT3活性而发挥作用[33]。当JAK/STAT3信号通路受到抑制时,可导致miR-17和miR-20a的过表达,相关分析还表明miR-17与JAK2表达密切相关[34]。另外,STAT3是miR-17的靶向分子,经双荧光素酶报告系统实验发现,miR-17的激活可以下调STAT3,p-STAT3和BCL2的表达而抑制人前列腺癌细胞(LNCaP)增殖并诱导细胞凋亡,其机制可能是miR-17与STAT3 mRNA 3′UTR结合而抑制其表达[35]。另外,有关miR-17-92簇干预骨骼肌分化的机制具有双向性,其中miR-17和miR-20a可以有效促进C2C12成肌细胞和原代细胞的分化,但miR-18a可能在C2C12细胞分化中起负作用[36]。因此,miR-17对肿瘤恶病质肌肉萎缩有抑制作用,其作用机制与抑制JAK/STAT3的表达密切相关。
1.4 JAK/STAT3促进肿瘤微环境生成加速肿瘤恶病质形成
JAK/STAT3信号传导对于微环境中的肿瘤-宿主相互作用非常重要。目前普遍认为,肿瘤微环境对肿瘤的生长和侵袭作用是不可或缺的[37]。JAK/STAT信号通路介导的STAT3激活在促进肿瘤细胞增殖分化和诱导侵袭中发挥重要作用,除充当信号传导和转录作用外,STAT3还可以通过表观遗传来激活相关基因和蛋白表达,促进上皮间质转化,对肿瘤微环境的形成有促进作用[38-39]。JAK/STAT信号通路参与机体免疫调节过程,比如STAT1和STAT2在抗肿瘤治疗中发挥重要的作用,其特异性表达可维持有效的免疫反应,但STAT3已被证实在肿瘤微环境中有参与癌细胞存活,调控免疫抑制,促进炎症反应的作用[32,40]。在头颈癌中,IL-6的过表达可通过激活JAK/STAT3介导上皮间质转化增加,导致细胞间连接和细胞极性的丧失,从而增加细胞迁移和侵袭性的可能性[41]。肿瘤的恶性程度与转移性病变的发展有关,涉及癌细胞从原发部位向远处器官扩散的复杂过程,肿瘤微环境(肿瘤细胞和周围组织)或大环境(包括循环中释放的肿瘤物质和其他组织分泌的促炎因子)在此期间发挥了关键作用,且这个发展过程会影响肿瘤相关恶病质进展[42],IL-6在肿瘤生长转移和恶病质的发展方面具有相关性,有研究者发现IL-6/JAK/STAT3前馈环驱动肿瘤发生和转移,调控肿瘤微环境的形成,并加速肿瘤恶病质的进程,尤其在肿瘤恶病质期间肌肉质量损失的调节[43-44]。由此可认为,JAK/STAT3的激活还可能通过影响肿瘤微环境导致肿瘤恶病质的进一步恶化。
2 药物干预JAK/STAT3信号通路减少肿瘤恶病质肌肉萎缩机制研究进展
2.1 JAK/STAT3相关抑制药物干预肿瘤恶病质肌肉萎缩机制研究
使用STAT3-SH2结构域模拟物抑制剂(SH2 domain-containing protein tyrosine phosphatase1,SPI)进行体外干预的实验表明,SPI可特异性的阻断STAT3的磷酸化进程,降低DNA结合活性和转录功能,广泛引起人乳腺癌、胰腺癌、前列腺癌和非小细胞肺癌的细胞产生形态变化,影响癌细胞的生存力和凋亡,并且该浓度下的SPI几乎不影响STAT1、STAT5或促丝裂原活化蛋白激酶途径的诱导和其它pTyr的形成,其机制可能是SPI充当STAT3-SH2结构域和pTyr相互作用的特异性抑制剂,同时SPI还破坏STAT3与IL-6R/gp130肽的结合[45]。鲁索替尼(ruxolitinib)磷酸盐是一种激酶抑制剂,可抑制janus相关激酶JAK1和JAK2,是第一个获准的专门治疗骨髓纤维化的药物,经美国食品与药物管理局批准上市,临床研究表明ruxolitinib不仅可有效改善原发性骨髓纤维化患者的输血需求和脾肿大症状,还可以增加患者体重,有效减缓体重耗损,使恶病质状态得到恢复,整体生活质量得到显著改善[46]。全身系统性炎症在肿瘤恶病质中普遍存在,炎症因子IL-6可激活UPS和JAK/STAT3信号通路[47-48],而ruxolitinib可能通过抑制炎症和JAK表达,进而干预肌肉萎缩进程[49]。舒尼替尼(sunitinib)是一类能够选择性地靶向多种受体酪氨酸激酶(receptor tyrosine kinase,RTKs)的新型药物,可用于治疗胃肠道基质肿瘤和转移性肾细胞癌,相关研究表明,sunitinib能够抑制STAT3和MuRF-1信号通路的过度激活,有效减缓肿瘤恶病质期间肌肉蛋白质分解代谢进程,并延长肾癌小鼠的生存期[50]。在一项研究中,真核生物起始因子4A(eukaryotic translation initiation factor 4A,eIF4A)抑制剂hippuristanol(Hipp)可显著降低IL-6分泌,降低p-STAT3蛋白表达,而对STAT3 mRNA水平没有影响,说明该抑制剂可能通过抑制eIF4A活性影响STAT3的翻译进而防止细胞因子诱导的肌肉消瘦[51]。泮托拉唑(pantoprazole)被定义为一种抗酸及抗溃疡药,但在一项恶病质小鼠实验中,pantoprazole以剂量依赖性方式降低血清IL-6和TNF-α的水平,表明pantoprazole减轻肿瘤恶病质所致肌肉萎缩的作用可能与其对炎症因子血清水平的调节有关,该实验还表明pantoprazole可作为JAK/STAT3途径的抑制剂,不仅有效降低p-STAT3表达水平,还通过抑制UPS降低MuRF1和Fbx32蛋白的表达,有效治疗肿瘤恶病质所致的肌肉萎缩和蛋白降解[52]。在患有胰腺癌的恶病质小鼠中,JAK2抑制剂AG490可以抑制JAK2的磷酸化和IL-6的表达水平,有效减缓体重下降进程[53]。C188-9是STAT3的小分子抑制剂,经C188-9治疗的C26肿瘤恶病质小鼠,抑制了STAT3的激活以及肌肉中C/EBPδ和UPS的表达,导致蛋白质合成增加和蛋白质降解减少,并增加小鼠体重,阻止肌肉质量耗损和肌肉纤维的减少[22]。
2.2 中药有效成分干预JAK/STAT3信号通路防治肿瘤恶病质肌肉萎缩机制
欧前胡素(imperatorin,IMP)是当归的主要生物活性成分,具有多种药理作用,如抗结肠炎,抗关节炎和抗肿瘤,研究表明,IMP可降低p-STAT3、MuRF1和Atrogin-1/MAFbx蛋白表达,是治疗肿瘤恶病质肌肉萎缩的潜在药物,其机制可能是IMP与STAT3的SH2结构域结合,进而抑制JAK/STAT3和UPS[54]。隐丹参酮是从丹参根中提取的主要亲脂性化合物,已被广泛用于治疗包括癌症在内的各种疾病,有数据显示,隐丹参酮可以抑制恶病质小鼠肌肉中MuRF1和MAFbx/Atrogin-1的表达,预防肿瘤恶病质体重减轻以及骨骼肌浪费,同时以剂量依赖性(2.5~10μmol/L)降低了C2C12肌管中MuRF1和MAFbx/Atrogin-1的表达,抑制STAT3转录活性,改善肌管萎缩,表明隐丹参酮可通过体内外途径抑制STAT3来防止肿瘤恶病质中的肌肉消瘦[55]。大蒜是常用和用途广泛的草药之一,研究者在大蒜中发现的一种硫化合物-Z-阿霍烯,其可以通过抑制炎症反应和肌肉蛋白降解以及促进肌肉蛋白质合成来减轻肿瘤恶病质所引起的骨骼肌萎缩,其机制可能是通过抑制JAK/STAT3途径并激活MAPK途径[56]。汇总见表1。
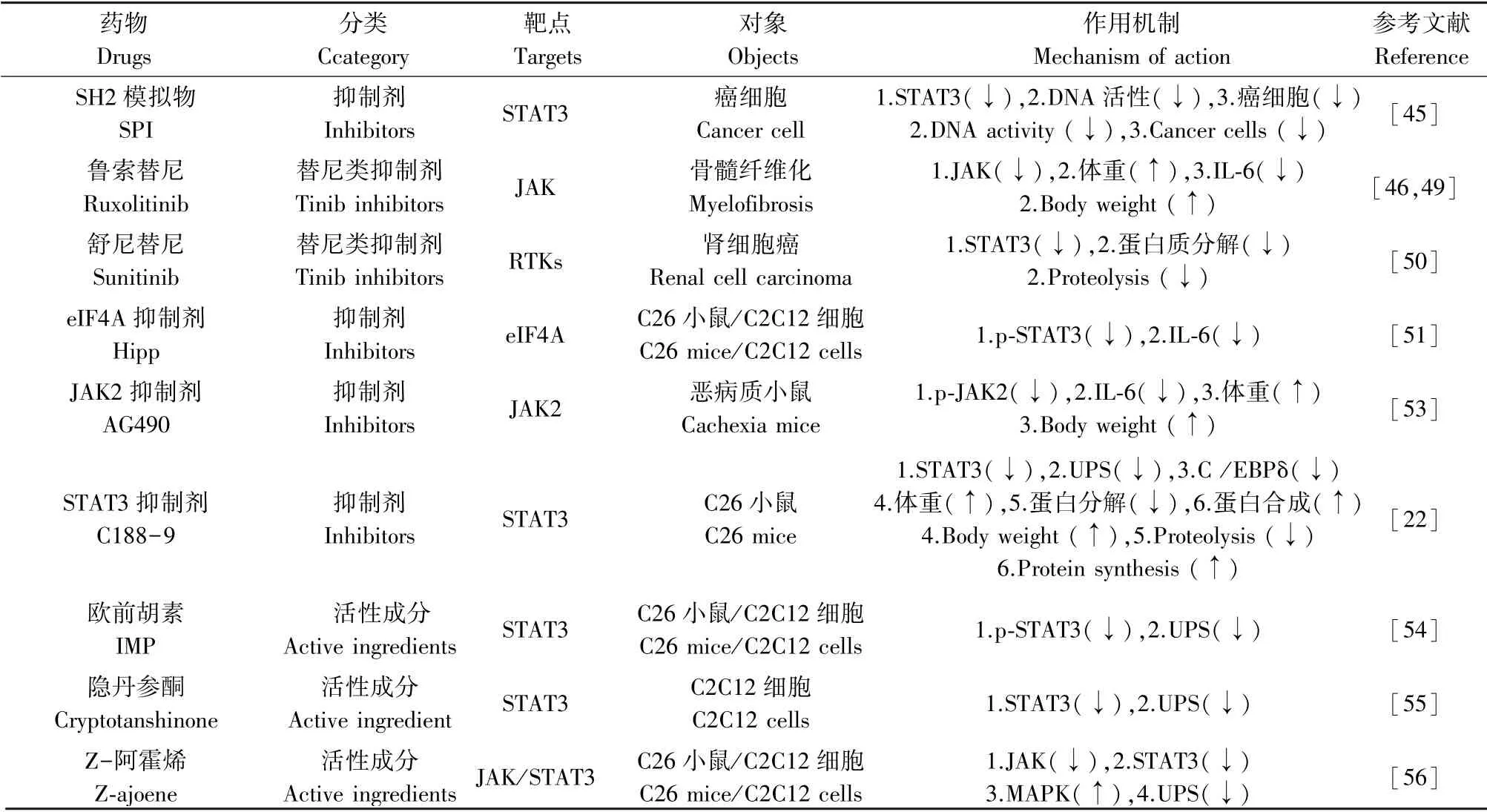
表1 药物干预JAK/STAT3信号通路介导肿瘤恶病质肌肉萎缩作用机制Table 1 Mechanism of drug intervention on JAK/STAT3 signaling pathway mediated muscle atrophy in cancer cachexia
3 总结和讨论
综上可知,JAK/STAT3信号通路可以从多个途径介导肿瘤恶病质肌肉萎缩,如通过调控泛素蛋白酶体系统加速骨骼肌蛋白质降解;通过调控自噬溶酶体系统引起肌肉萎缩,另外microRNA、肿瘤微环境与JAK/STAT3信号通路的相互作用可诱导肿瘤细胞的增殖、迁移、侵袭,加速肿瘤恶病质的发生发展。基于以上机制,本文还分析相关药物干预信号通路的临床和基础研究,结果表明JAK/STAT3相关抑制药物可在体内外抑制JAK、STAT3、p-STAT3、IL-6和TNF-α等的表达,并进一步抑制UPS,相关的靶向干预在临床和基础研究中均有显著的作用,可有效抑制肌肉蛋白降解,减缓肌肉萎缩进程,保持体重,改善晚期肿瘤恶病质患者生活质量。另外一些中药有效成分如IMP、隐丹参酮、Z-阿霍烯也可以通过干预JAK/STAT3信号通路防治肿瘤恶病质肌肉萎缩。
尽管大量的体内外实验和临床观察表明抑制JAK/STAT3信号通路可有效减缓肿瘤恶病质肌肉萎缩进程,然而目前仍没有一种专项治疗肿瘤恶病质的药物批准上市,一些靶向抑制JAK的上市药物如Ruxolitinib、Tofacitinib,其主要临床应用也并不是用于治疗肿瘤恶病质。另外造成肿瘤恶病质发生的因素除大量蛋白质耗损引起的骨骼肌萎缩外还与厌食症、中枢神经系统、脂肪代谢异常、线粒体能量代谢异常、肝代谢异常[13]密切相关,这使得该综合征具有复杂性和多样性,为治疗肿瘤恶病质肌肉萎缩增加更多的不确定因素。为了更好的认识肿瘤恶病质肌肉萎缩的机制,早日为肿瘤晚期恶病质患者保持体重、延长生存期提供有效药物,针对JAK/STAT3通路介导肿瘤恶病质肌肉萎缩机制仍需进一步研究同时应对现有的信号通路抑制剂在治疗肿瘤恶病质肌肉萎缩方面开展更多与药效、安全性相关的基础研究和临床观察。
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31 09:59:20
健康护理(2022年3期)2022-05-26 02:30:19
中学生数理化·七年级数学人教版(2019年10期)2019-11-25 07:33:58
中学生数理化·高一版(2018年9期)2018-10-09 06:46:50
湖南教育·C版(2018年3期)2018-06-05 16:54:36
中国运动医学杂志(2016年3期)2016-07-10 12:07:23
中国运动医学杂志(2016年3期)2016-07-10 12:07:23
医学研究杂志(2015年5期)2015-06-10 06:43:26
中国肿瘤临床(2014年14期)2014-01-24 02:54:13
中华结直肠疾病电子杂志(2014年5期)2014-01-22 07:59:16
