
一种基于高通量测序的拷贝数变异检测自动化分析解读及报告系统
2021-08-19 08:13蔡瑞琨曹宗富喻浴飞陈翠霞张钏罗敏娜李乾马旭
生殖医学杂志 2021年8期
蔡瑞琨,曹宗富,喻浴飞,陈翠霞,张钏,罗敏娜,李乾,马旭*
(1.国家卫生健康委科学技术研究所,北京 100081;2.国家人类遗传资源中心,北京 102206;3.甘肃省妇幼保健院医学遗传学中心,兰州 730050)
基因组拷贝数变异(Copy number variation,CNV)是染色体结构变异的一种,通常是指在基因组上长度在1 kb以上的大片段碱基序列的增加或者减少,主要表现为亚显微水平的缺失或者重复。它与单核苷酸多态性(Single nucleotide polymorphism,SNP)同样在人群中普遍存在,是人类基因组水平上广泛分布的一种变异形式[1-2]。近年来,随着基因组学实验技术的迅猛发展,高通量测序技术为研究基因组水平上的变异提供了强有力的工具[3]。与疾病相关的基因组水平上的变异研究,不仅仅局限在SNPs,CNVs也可以通过改变基因的倍数、打乱基因的结构等方式影响个体的表型,从而导致单基因遗传病和复杂疾病[4]。例如,在神经发育类疾病的研究中发现CNVs是一个重要的危险因素[5],在一些散发病例中可以发现共同的新生罕见CNV[6]。CNVs在高危妊娠、自然流产以及遗传性疾病中有比较高的检出率,提示在此类人群中CNVs检测的重要性[7]。在癌症研究方面,CNVs被认为是一种潜在的肿瘤诊断生物标记物,多种癌基因都与CNVs相关,因此检测不同肿瘤的CNV具有重要意义[8-9]。
随着人类基因组学和疾病组学的发展,人类医学正逐步迈入到精准医学的时代,可以根据个体的遗传学背景,阐明个体疾病的发病原因、预测发病风险,以及进行个性化的健康管理和治疗。这对个体基因变异的检测提出了更高的要求,不仅仅满足于SNPs的检测,还包括了CNVs的检测和分析解读。而CNVs检测应用于精准医学方面所面临的挑战在于:(1)复杂的分析流程。从原始的高通量测序数据,到分析出CNVs,中间需要若干分析步骤,运行多种分析软件,不仅消耗大量时间,且技术门槛较高,很难为广大非生物信息学人员所使用。(2)致病变异的鉴定和解读。对检测出的众多CNVs进行筛选,评价其与表型的关联,需要消耗大量时间、查阅大量文献和数据库,逐个审核和确认,效率非常低[6]。为此,我们基于已有的单基因病遗传变异解读系统,针对CNVs的分析流程,又开发出一套基于云的可视化自动化智能化的CNVs变异检测分析解读系统,以满足单基因病和复杂疾病在CNVs检测方面的临床和科研需求,并通过https://www.pgenomics.cn/提供免费的分析解读服务。
资料和方法
一、原始数据的预处理
分析流程处理的原始数据来自于高通量测序技术产生的全基因组测序数据或者外显子组测序数据。在Linux系统环境中,首先使用FastQC软件对fastq格式的原始测序数据进行质量控制。然后,将质量过关的数据用BWA MEM软件[10]与人类参考基因组(hg19版本)进行序列比对,并用Samtools软件[11]得到比对后的Bam格式文件。
二、基于对照样本数据建立参考基线
将输入的与待测样本同批次的正常样本做对照,建立一个参考基线。分别计算每个对照样本的目的区域内和目的区域外的测序深度,合并所有对照样本,矫正GC含量等系统误差,构建正常人样本的基因组的测序分布模型;在Linux系统环境中,对于全基因组测序数据采用CNVKit软件[12]实现,对于全外显子测序数据采用ExomeDepth软件实现。
三、变异的检测
将待测样本与对照样本建立的参考基线做比对,检测出待测样本中相应的变异情况。分别计算每个待测实验样本的目的区域内和目的区域外的测序深度,然后计算它们相对于对照样本的log2 ratio值,再进行小片段划分,并计算每个小片段区域的绝对拷贝数。在Linux系统环境中,对于全基因组测序数据采用CNVKit软件实现,对于全外显子测序数据采用ExomeDepth软件实现。
四、变异的注释
对识别的CNVs,根据公共数据库对变异起始/终止位置、所覆盖的基因、具体的变异类型、在世界不同人群中的频率,以及DGV数据库[13]、千人数据库、dbVar数据库和OMIM数据库中已知变异的致病情况等进行注释。在Linux系统环境中采用AnnotSV软件进行注释,并且该软件还使用了美国医学遗传学和基因组学学院(ACMG)定义的分类标准,给出了初步的致病性分类。
五、变异的致病性评分
根据变异注释结果中分析软件注释出的变异信息与用户提交的相关信息之间的近似程度等情况对结构变异进行累计评分,最终的变异评分定义为多种注释结果的加权评分之和,具体方法为
其中,wi为不同证据的评分权重,si为每个证据的评分;wi默认值为1,可根据情况进行调整。其中,证据评分包括以下几项:(1)在注释结果中给出的变异初步致病性分级;(2)用户输入的疾病名称与注释结果中分析软件注释出的此变异对应的疾病名称,两个疾病名称之间的匹配程度;(3)疾病的已知致病基因加权评分;(4)对用户输入的表型和每个结构变异片段注释的表型,两个表型之间利用多层级聚类算法计算两者匹配相似度分值;(5)各变异数据库中不同人群的最大频率MAX_AF情况;(6)变异所在位置的重要性。在Linux系统环境中使用python语言开发完成此功能模块。
六、变异的自动化推荐以及可视化
由于上述分析软件在Linux系统下基于命令行模式下运行,用户友好度低,因此,本研究集成了各应用软件,采用友好的流程管理方案,使用户可以直接使用Windows系统通过Web浏览器直接访问并分析数据,实现了数据的自动化分析和结果的可视化展示。最终的分析报告及可视化页面直接在Web浏览器中展示,根据变异的总评分降序和基因名称升序对变异进行排序。变异的总评分越高,说明变异与用户提交的疾病名称或表型越匹配,且致病性越高,从而实现了致病变异的推荐功能;推荐页面包括了变异的位置、覆盖的基因名称、变异评分分值、染色体水平上突变的位置图,以及相关的表型信息和变异频率信息等。
结 果
我们基于云开发了可视化自动化智能化的CNVs变异分析解读及推荐系统,实现从新一代测序原始数据到致病CNVs推荐的自动化流程(图1),极大降低了CNVs变异数据分析工作和人工解读的工作量,大大提高了CNVs分析和临床解读的效率。通过https://www.pgenomics.cn/提供免费共享服务,用户注册申请后即可登录使用。

图1 拷贝数变异检测自动化分析流程图
用户可根据自己的实验方案和已有的测序数据选择对应的分析流程,主要分为:基于全基因组的CNV测序(CNV-seq)数据的分析流程,和基于全外显子组测序数据的分析流程。分析流程导入的数据可以是测序的原始fastq文件格式,也可以是分析过程中的bam文件格式。根据实验方案,可以选择使用用户自己的对照样本作为检测CNV的参考基线,也可以使用平台上提供的参考基线。
示例为一位有磁共振磨牙征等表型的患者的拷贝数分析结果(图2)。在提交分析流程前,用户需要输入数据对应的患者的疾病名称或者HPO标准表型。例如,此示例输入的标准表型为“HP:0002419磁共振磨牙征;HP:0001510生长延迟;HP:0007033小脑发育不良;HP:0000639眼球震颤”。提交分析后,系统会自动进行分析,给基因和变异进行评分,与标准表型相关性高的将会赋予更高的分值。待分析结束后,即可查看报告。报告页面的左侧显示分析的流程,中间部分则是每个基因拷贝数的分析结果,根据拷贝数评分的分值从大到小排序,展示出拷贝数、评分分值、区域范围、范围内覆盖的基因名称、相关的疾病名称、遗传模式和拷贝数的长度等信息。此示例推荐第一位的是TMEM237基因,基因对应的疾病为Joubert综合征14型,变异区域为基因的1号外显子区域到2号内含子区域,对应的OMIM编号为614423,平台附有链接,可以直接点击进入OMIM数据库进行查看。
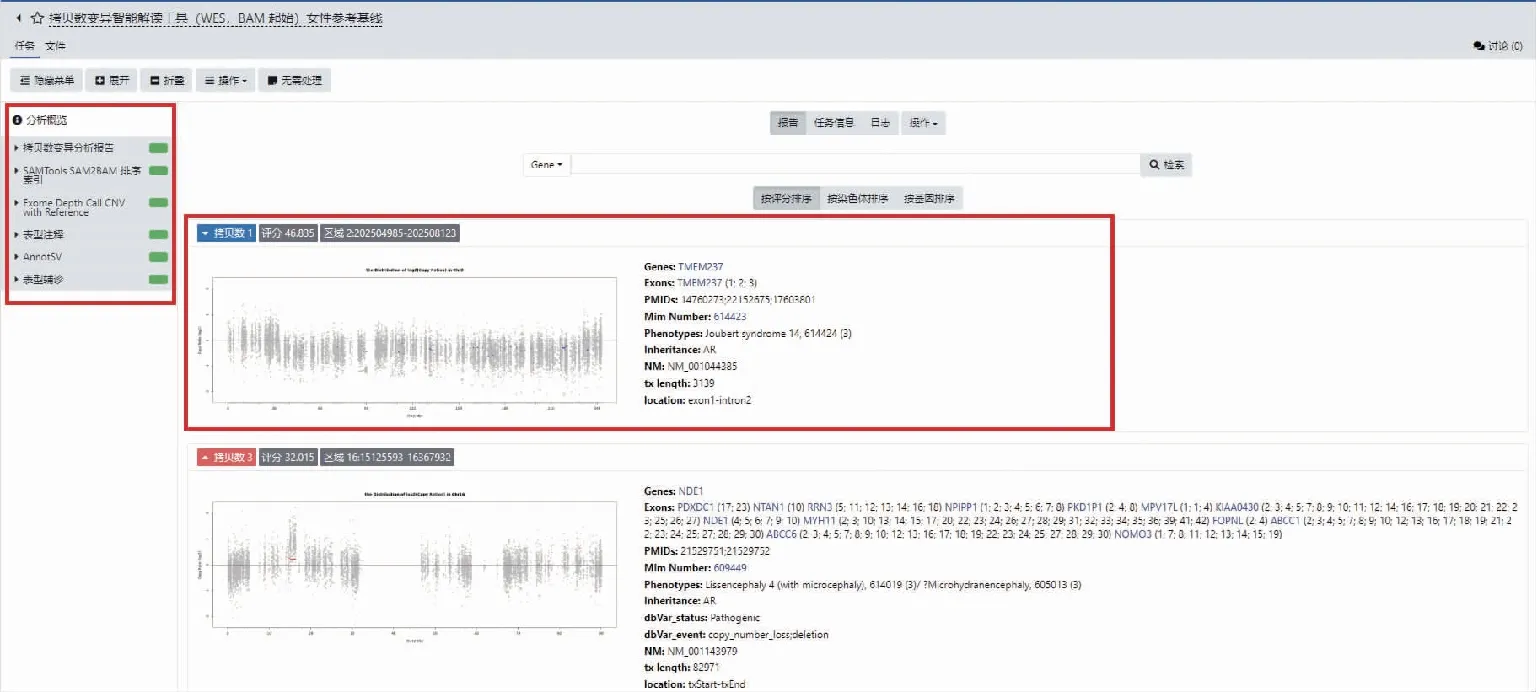
图2 拷贝数分析流程报告界面
讨 论
本研究基于高通量测序数据,包括全基因组测序数据和外显子组测序数据,面向遗传病和肿瘤两大领域中的拷贝数变异致病性研究,可与SNV/Indel变异致病性研究相结合,共同研究疾病的发病机制、病因诊断,以及产前遗传病诊断和筛查[14]。
在技术上,本研究采用了生物信息学技术,集成了多个CNV分析相关的应用软件,并且开发出致病突变的推荐功能模块,使得CNV分析流程从原始数据处理到最终的致病变异推荐功能全部一次性自动化地分析完成。本系统在功能上实现了一种全自动可视化的拷贝数变异检测和推荐,能够对高通量测序的原始数据进行分析,检测出其中的拷贝数变异,并根据数据对应的临床表型进行判读,结合拷贝数变异公共数据库的注释信息,对变异进行综合评分和致病性分级,最终推荐和报告检测个体中的致病突变。现有分析软件多是Linux系统下命令行实现,本系统面向广大无生物信息学背景的临床医生和科研人员,整个分析流程用Windows系统的Web浏览器即可访问使用,并将分析结果进行了可视化展示,真正地实现了拷贝数变异检测的全程自动化可视化检测,加快了研究和诊断速度,极大地节约时间和人力成本。并且,本系统已经面向多个省市的多家临床医疗机构和科研院所的遗传领域临床医生和科研人员免费开放使用[15-16]。
猜你喜欢
右江民族医学院学报(2022年2期)2022-05-19
河北果树(2021年4期)2021-12-02
河北医学(2021年10期)2021-10-27
天津医科大学学报(2021年1期)2021-01-26
医药前沿(2020年20期)2020-11-10
趣味(数学)(2020年4期)2020-07-27
支部建设(2020年15期)2020-07-08
中国临床医学影像杂志(2019年6期)2019-08-27
河北农业科学(2019年6期)2019-03-21
百科知识(2015年18期)2015-09-10
