
乙烯原位共聚制备线性低密度聚乙烯的双功能催化剂体系研究进展
2021-08-13 08:48谢光勇秦亚雯罗德荣李建蒙祥
中南民族大学学报(自然科学版) 2021年4期
谢光勇,秦亚雯,罗德荣,李建,蒙祥
(中南民族大学 化学与材料科学学院 & 催化转化与能源材料化学教育部重点实验室,武汉 430074)
聚烯烃材料是产量最大、应用最广的一类合成树脂,烯烃聚合催化剂是决定聚合物性能的关键因素,因此催化剂的研发是核心和动力.目前,烯烃聚合催化剂的发展经历了Ziegler-Natta催化剂、茂金属催化剂、非茂过渡金属催化剂三个阶段,开发出更高效并且聚合物结构更可控的烯烃聚合催化剂一直是高分子学科领域的研究热点[1-2].
聚乙烯(PE)作为最常用的聚烯烃材料,2020年的产量预计将达到1.08亿t,而占有聚乙烯市场40%以上的线性低密度聚乙烯(LLDPE),由于其高撕裂强度和抗冲击强度,成为全球增长最快的通用塑料.LLDPE分子结构呈带有一定长度支链的线性,密度约为0.93 g·cm-3,LLDPE较普通聚乙烯性能更加优异,撕裂强度、抗穿刺强度、拉伸强度与断裂伸长率均可大幅提升,广泛应用于薄膜、管材、包装和电线电缆等领域.
LLDPE一般先通过乙烯齐聚获得α-烯烃,α-烯烃分离提纯后再与乙烯共聚得到,即所谓的两步法.1984年BEACH[3]和KISSIN[4]首次提出了双功能催化体系催化乙烯原位共聚制备LLDPE,即将齐聚和共聚催化剂加入同一聚合装置中,乙烯先在齐聚催化剂上制得α-烯烃,随后α-烯烃立即与过量乙烯通过共聚催化剂制得LLDPE(图1).这种一锅法与分步法相比,最显著的优点是单体原料只有乙烯一种,共聚单体α-烯烃由体系中齐聚催化剂所得,省去了α-烯烃的合成、提纯和储存等步骤,生产工艺更加简捷,大大降低了生产成本,因此备受关注.

图1 双功能催化剂催化乙烯原位共聚制LLDPEFig.1 Bifunctional catalysts for ethylene in-situ copolymerization to LLDPE
双功能催化体系中催化剂的选择是关键,首要条件是各催化剂和助催化剂之间没有毒化作用导致失活,并且匹配好齐聚反应和共聚反应的速度,使聚合反应平稳可控地进行.双功能催化剂体系既可以是2~3种分别拥有齐聚或共聚活性的催化剂的混合,也可以将齐聚和共聚活性中心构建于同一个结构中.由于条件比较苛刻,目前有关双功能催化剂体系的文献并不多,本文综述了近年乙烯原位共聚制备LLDPE催化剂的研究进展,主要对催化剂的组成、结构、助催化剂种类和聚合物结构进行论述.
1 双功能催化剂体系
在双功能催化剂体系中,齐聚催化剂一般选用铁系、铬系或钛系配合物;根据组成双功能催化剂体系中共聚催化剂结构差异,将其划分为Ziegler-Natta催化剂、茂金属催化剂和非茂金属催化剂三种体系.
1.1 Ziegler-Natta催化剂作共聚催化剂
20世纪50年代Ziegler-Natta催化剂的出现推动世界塑料工业发展产生深刻变革.BEACH等[3-4]首次以δ-TiCl3·0.33 AlCl3、TiCl4/MgCl2/PE或VOCl3为共聚催化剂,钛系、镍系为齐聚催化剂,组合成用于乙烯原位共聚的双功能催化剂体系.该催化体系所制备的LLDPE具有很高的1-丁烯插入率,支化度最高可达45(1000 C),但该催化体系中各催化剂之间的干扰较大,齐聚催化剂的比例增加会使得活性降低.
吡啶二亚胺铁、钴配合物1[5]在乙烯齐聚中表现出很好的催化活性和选择性(图2),因此是双功能催化体系中理想的齐聚催化剂.LU等[6]以吡啶二亚胺铁配合物(1a)与传统的Ziegler-Natta催化剂TiCl4/MgCl2组成双功能催化剂体系,甲基铝氧烷(MAO)和三乙基铝(TEA)为助催化剂,成功催化乙烯原位聚合制备支化聚乙烯.通过调节TEA和MAO用量、铁催化剂与Ziegler-Natta催化剂比例以及反应温度,可有效调控支化度分布范围在8~29(1000 C),产物中支链类型丰富,包括乙基和丁基短支链以及C6以上长支链;该产品具有比茂金属催化剂更高的分子量和更宽的分子量分布,为材料提供了优异的机械性能和加工性能.
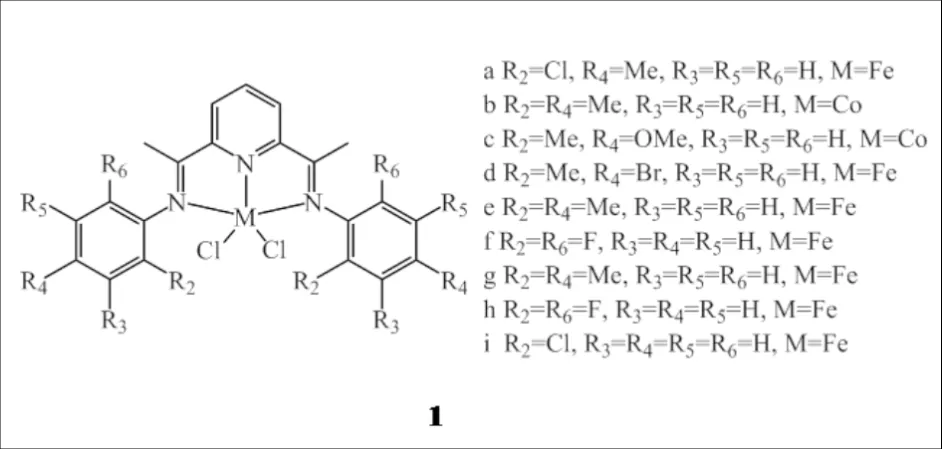
图2 吡啶二亚胺铁、钴配合物1Fig.2 Bis(imino)pyridyl iron/cobalt complexes 1
SNS-Cr配合物2[7]是一类极具发展潜力的乙烯齐聚催化剂(图3),具有优异的催化活性、选择性和热稳定性,对1-己烯的选择性基本在99%以上.郭松[8]以高活性、高选择性及副产物少的SNS-Cr配合物2g为乙烯三聚催化剂,Ziegler-Natta催化剂为共聚催化剂,选用TEA/MAO混合助催化剂体系,成功催化乙烯合成出含丁基支链的线型低密度聚乙烯,产物熔点为122.3 ℃并且呈现出较好的颗粒形态.配合物2g可以在较少助催化剂用量下保持较高的反应活性和选择性,并且增加配合物2g的比例会降低所得聚合物的分子量和熔点,结晶度约为50%~60%.
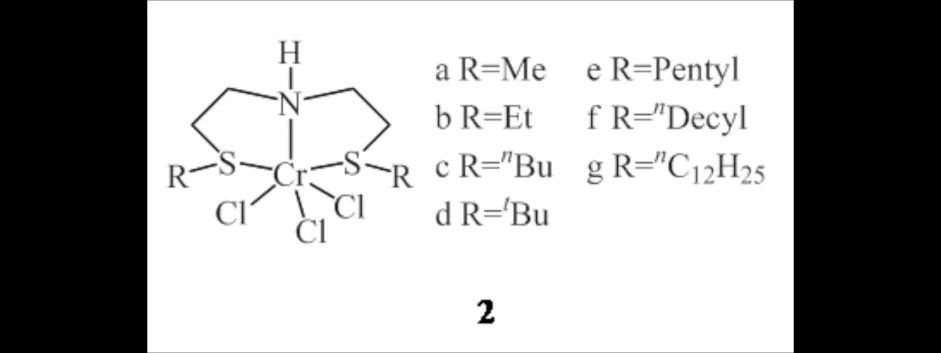
图3 SNS-Cr铬配合物2Fig.3 SNS-Cr chromium complexes 2
Ziegler-Natta催化剂作双功能催化剂体系的共聚催化剂时,通常需要MAO/TEA作混合助催化剂才具有更好效果,可能会产生两种助催化剂相互干扰的难题;此外,Ziegler-Natta催化剂中含有多个活性中心,因此制得的聚合物结构不稳定,不利于催化剂的研究,导致其在原位共聚中的应用受限.
1.2 茂金属作共聚催化剂
当选用MAO作为助催化剂时,茂金属催化剂具有很高的活性和选择性,并且制备的聚合物具有较窄的分子量分布,这些显著优点使其在烯烃聚合催化剂领域受到较多的关注.因此现主要选用茂金属催化剂作为乙烯原位共聚制备线型低密度聚乙烯的双功能催化剂体系中的共聚催化剂.
1.2.1 与乙烯二聚催化剂组成双功能催化剂体系
BAMHART等[9-10]以具有优异催化性能的限定几何构型共聚催化剂CGC-Ti(3)分别与齐聚催化剂锆(4)或镍配合物(5)组成双功能催化剂体系(图4),实现乙烯原位聚合得到乙基支链为主的聚乙烯.Ti(3)-Zr(4)体系均以MAO为助催化剂,避免了因助催化剂不同而导致的催化剂之间的相互干扰,但选择性方面齐聚催化剂对乙烯二聚表现较差,除了主产物1-丁烯外,还会产生含量较高的不能共聚插入主链的副产物2-烯烃、2-烷基-1-烯烃等.但当Ti(3)-Ni(5)体系以B(C6F5)3为助催化剂时,1-丁烯的选择性在90%以上,并且当n(Ti)/n(Ni)<1时,还会产生部分1-己烯.
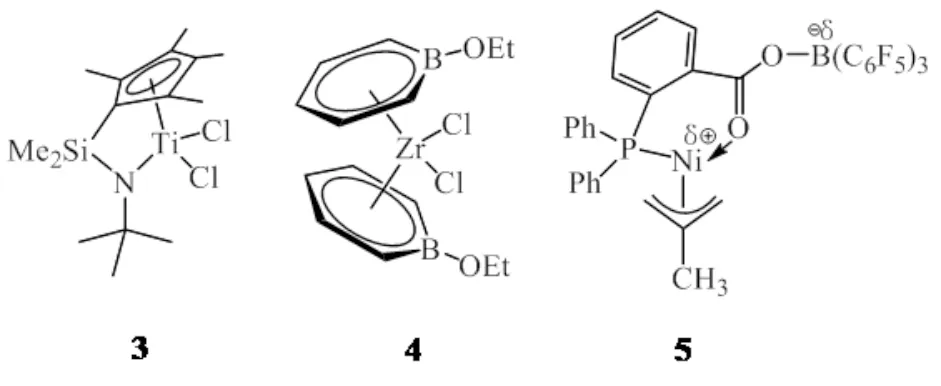
图4 钛、锆和镍配合物3~5Fig.4 Titanium, zirconium and nickel complexes 3-5
FREDIANI等[11]以吡啶亚胺钴齐聚催化剂(6, 图5)和CGC-Ti(3)组成原位共聚催化剂体系,MAO做助催化剂,在30 ℃和3 atm乙烯压力下聚合1 h后,齐聚催化剂6对1-丁烯表现出80%以上的选择性,催化活性最高可达106g·mol-1·h-1,吡啶环上R基团位阻增大会使聚合物分子量分布和支链度增加.
SÉMERIL等[12-13]以经典的共聚催化剂Cp2ZrCl2分别与钴催化剂(6)或大环结构镍催化剂(7)组成双功能催化剂体系(图5),该体系在助催化剂MAO作用下均能在乙烯原位共聚中表现出高活性.值得注意的是具有特殊的大环结构和配位环境的镍催化剂7可以在P—Ni—P处形成周期性摆动,当其键角运动到最大值时,活性达到最高,对1-丁烯的选择性也在95%以上.

图5 钴配合物 6和镍配合物 7Fig.5 Cobalt complexes 6 and nickel complex 7
1.2.2 与乙烯三聚催化剂组成双功能催化剂体系
含有丁基、己基等长支链的线型低密度聚乙烯具有优良的机械与加工性能,因此提高双功能催化剂体系中乙烯齐聚选择性和单体插入率,是乙烯原位共聚催化剂研究的重要方向.
YE等[14]以乙烯三聚选择性高达80%(摩尔分数)的单茂钛配合物8(图6)和CGC-Ti(3)组成双功能催化剂体系,在助催化剂改性甲基铝氧烷(MMAO)作用下催化乙烯原位聚合得到只含丁基支链的聚乙烯;聚合物支化度受催化剂比例和温度影响,最高达40(1000 C),产物具有典型的流变性能,如增强的零剪切粘度、更高的动态模量和热流变学复杂性等.

图6 单茂钛配合物8Fig.6 Mono-metallocene titanium complex 8
SNS-Cr(2)作为一系列活性高、对1-己烯选择性好的齐聚催化剂,也常常应用于双功能催化体系中.WET-ROOS等[15]以乙烯三聚催化剂SNS-Cr(2f)分别与CGC-Ti(3)、Cp2ZrCl2及Me2Si(2-Me-Ind)2ZrCl2(9,图7)组成双功能催化剂体系,MAO做助催化剂,结果表明共聚催化剂的不同对1-己烯的插入影响较大,其中Me2Si(2-Me-Ind)2ZrCl2最高,达5.81%,Cp2ZrCl2最低,仅为0.65%;聚合物中只有丁基支链,表明3种催化剂对1-己烯都具有很高的选择性,这种高选择性使产品与纯的乙烯/1-己烯共聚物性能非常接近,较具工业应用价值.
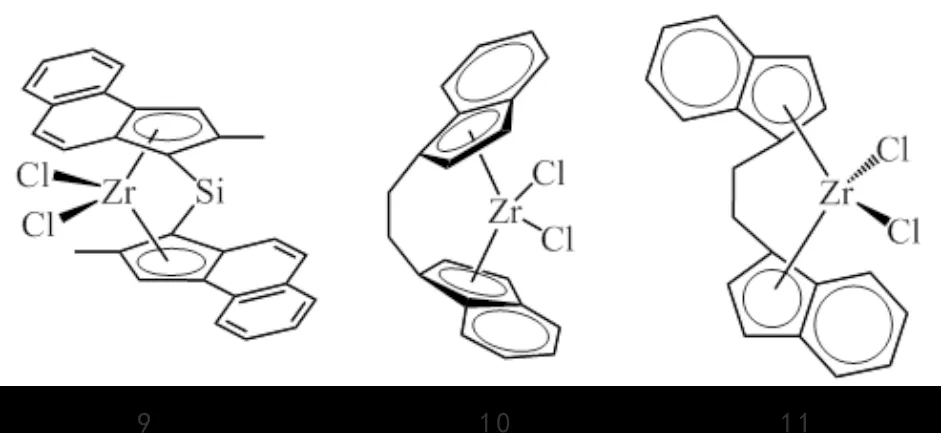
图7 具有共聚活性的锆配合物9~11Fig.7 Zirconium complexes 9-11 with copolymerization activity
ZHANG等[16-17]结合动力学模型对SNS-Cr催化剂(2b,2g)与两种不同的共聚催化剂CGC-Ti(3)和Et(Ind)2ZrCl2(10,图7)组成的双功能催化剂体系进行了研究,在半连续反应器中成功制备丁基支链聚乙烯.结果表明:齐聚反应速度与共聚反应速度之间存在差异,可以通过调节催化剂比例、聚合温度、助催化剂用量、聚合压力和预齐聚时间等条件有效调控聚合物结构和产品性能;该方法还制备了1-己烯插入量达12.0%(摩尔分数)的聚烯烃弹性体POE,与相似熔点和结晶度的DOW产品8150相比,其断裂强度、断裂伸长率和弹性恢复能力较好.
MOHAMADNIA等[18]以戊烷基取代的SNS-Cr(2e)与外消旋的rac-Et(Ind)2ZrCl2共聚催化剂(11,图7)组成乙烯原位共聚催化剂体系,MAO为助催化剂,体系中齐聚催化剂对乙烯三聚选择性高达99.9%,所得聚合物以丁基支链为主,支链度可达17.1(1000 C).
MOHAMADNIA等[19]以丁基取代的SNS-Cr为齐聚催化剂,Cp2ZrCl2为共聚催化剂,在MMAO作用下催化乙烯原位聚合得到丁基支链聚乙烯.催化活性、聚合物结晶度、支化度等性能受到催化剂比例Cr/Zr的较大影响,当Cr/Zr摩尔比为2时,支化度高达41.52(1000 C).另外,通过在聚合过程中添加TiO2、SBA-15和磁性Fe3O4纳米颗粒填料,可制备具有亲水性的线性低密度聚乙烯纳米复合材料,TiO2型纳米复合材料对革兰氏阴性细菌还具有抗菌活性.
SUZUKI[20]和SATTLER等[21]的研究表明:非茂类过渡金属钛配合物12(图8)在助催化剂MAO作用下能高活性高选择性催化乙烯三聚得到1-己烯,选择性高达95.4%,当R为位阻较大的金刚烷基(12c)时可显著提高选择性和催化活性.
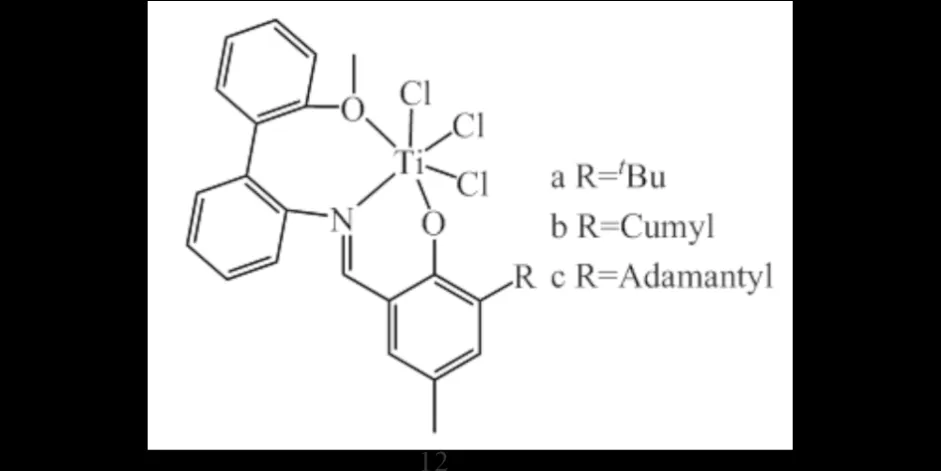
图8 [ONO]TiCl3齐聚催化剂12Fig.8 [ONO]TiCl3 oligomerization catalysts 12
KARBACH等[22]以取代基为金刚烷的钛配合物12c与Cp2ZrCl2组成双功能催化剂体系,将两种配合物分别负载在硅胶载体上,可以解决两个不同的金属活性中心之间的相互影响的问题,达到更好的催化效果,成功催化乙烯原位共聚制备乙烯/1-己烯共聚物.
ALUTHGE等[23]以金刚烷取代的[ONO]三齿钛配合物12c为齐聚催化剂,共聚催化剂则为CGC-Ti,由于是相同的钛金属中心,故可以将两种催化剂负载在同一硅胶载体上;产物中的丁基支链含量可以通过聚合条件有效调控,使用不同的助催化剂(MAO或MMAO)也会产生不同的乙烯三聚中间体[24].
1.2.3 与乙烯四聚或齐聚催化剂组成双功能催化剂体系
共聚物中己基等长支链的含量对材料的性能具有较大影响,但1-辛烯的合成尚未实现工业化,其主要从乙烯的齐聚产物中分离得到.因此,设计出能催化乙烯原位生成1-辛烯的双功能催化体系是制备含长支链线型低密度聚乙烯的可行办法.
JIANG[25]和WET-ROOS等[26]以对乙烯四聚具有高活性高选择性的[PNP]三齿铬配合物13为齐聚催化剂[27],rac-Et(Ind)2ZrCl2(11)和Ph2C(Cp)(9-Flu)ZrCl2(14)为共聚催化剂(图9),MAO为助催化剂,催化乙烯原位共聚成功制备了LLDPE;rac-Et(Ind)2ZrCl2所得聚合物中支链丁基和己基含量为0.43%和1.02%,而Ph2C(Cp)(9-Flu)ZrCl2所得聚合物支化度更高,1-己烯和1-辛烯的插入率达0.6%和1.5%.聚合物中己基支链的含量可通过催化剂配比、助催化剂用量和反应温度等聚合条件有效调控,进而改善共聚物的性能.

图9 [PNP]铬配合物13和茂锆配合物14Fig.9 [PNP] chromium complexes 13 and zirconocene complex 14
SCHWERDTFEGER等[28-29]以叔丁基取代的SNS-Cr配合物2d为齐聚催化剂,锆配合物15为共聚催化剂(图10),在MAO的作用下,制备的LLDPE结构以己基及更长支链为主,同时含有少量乙基和丁基支链.改变助催化剂的用量可有效调控1-己烯、1-辛烯的含量,其插入率在较长的聚合时间下会有所提高,催化剂配比也会影响支化度,聚合物中长支链的形成可通过“链行走机理”解释.
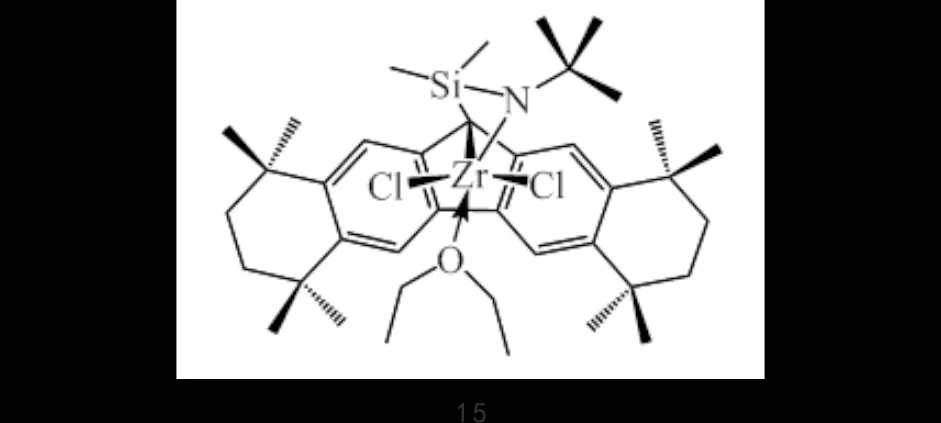
图10 锆配合物15Fig.10 Zirconium complex 15
KOMON等[30]以两种镍齐聚催化剂16和17(图11)与CGC-Ti(3)共同组成三元的双功能催化剂体系,在助催化剂B(C6F5)3作用下得到多种支链类型的聚乙烯.该体系中,PO^Ni(16)主要催化乙烯二聚产生1-丁烯,而NN^Ni(17)主要得到1-己烯、1-辛烯等长链α-烯烃,并且均对乙烯齐聚表现出较高的选择性,改变两种镍催化剂的配比可有效调控线型低密度聚乙烯中的支链结构及分布.
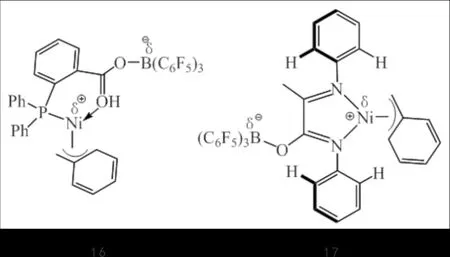
图11 镍配合物16和17Fig.11 Nickel complexes 16 and 17
ZHANG等[31]以苯氧亚胺型FI-Zr配合物为齐聚催化剂,CGC-Ti为共聚催化剂,成功合成了具有结晶长支链的新型梳形乙烯/1-辛烯共聚物弹性体(CPOE).FI-Zr合成具有超过88%带末端双键的线性PE大分子单体,而CGC-Ti用于乙烯、1-辛烯和PE大分子单体的共聚合;掺入超过20%(摩尔分数)的1-辛烯可确保CPOE的无定形主链,得到的CPOE同时具有高熔融温度(>120 ℃)和低玻璃化转变温度(< -60 ℃),其断裂伸长率大于1200%,部分样品比商业POE Engage 8150表现出更好的应变恢复.
ZHANG等[32]以吡啶二亚胺铁(1b)为齐聚催化剂与锆配合物Et(Ind)2ZrCl2(10)组成双功能催化剂体系,MAO作助催化剂,可得到长支链的LLDPE.该催化体系助催化剂用量少、催化活性高、热稳定性好,并且所得聚合物的结构和性能可通过各种催化条件进行调控;聚合活性最高达1.2×107g·mol-1·h-1,80 ℃时仍具有6.9×106g·mol-1·h-1的催化活性,低温聚合所得产物为双峰分布,而80 ℃时呈现单峰.
QUIJADA等[33]选用乙基取代的吡啶二亚胺铁配合物(1c)作为齐聚催化剂,将其分别与具有共聚催化性能的锆配合物Et(Ind)2ZrCl2(10)或Me2SiInd2ZrCl2(18,图12)组合,在助催化剂MAO的作用下,2 atm乙烯压力时原位聚合得到支化度分别达20(1000 C)和14(1000 C)的丁基支链LLDPE;体系中齐聚催化剂与共聚催化剂的比例增加会使催化活性和聚合物分子量降低;在相同的催化条件下,1c/18双功能催化剂体系制得的产物拥有更宽的分子量分布和更高的支化度,但1c/10催化剂体系可以得到更高的催化活性和聚合物分子量.
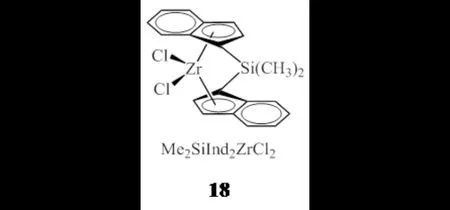
图12 锆配合物18Fig.12 Zirconium complex 18
1.2.4 桥联双功能催化剂体系
与组合型的双功能催化剂体系不同,KUWABARA等[34]通过化学键将具有共聚性能的前过渡金属茂锆和具有齐聚性能的非茂类后过渡金属构建于同一配合物中得到异双核配合物19(图13),配合物兼具齐聚和共聚活性中心,在助催化剂MMAO作用下,成功催化乙烯原位聚合得到含乙基和C6以上的长支链聚乙烯.与对应的独立两个单核配合物组成的混合催化体系相比,所得的聚合物支链度更高,分子量分布更窄.
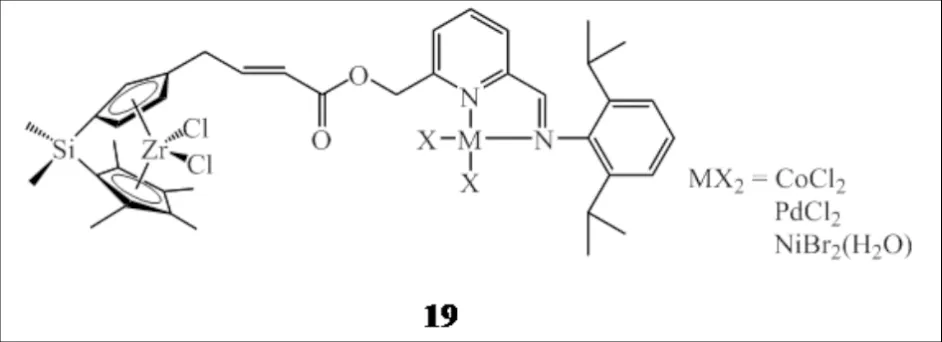
图13 桥联双功能催化剂19Fig.13 Bridged bifunctional catalysts 19
MARKS等[35-37]接连报道了两种以烷基桥联的异双核配合物,即CGC2-TiZr配合物20和{Ti-Cr}配合物21(图14). CGC2-TiZr配合物20中,锆金属中心作为齐聚活性中心产生1-己烯及更长链的α-烯烃,钛中心作为共聚活性中心,成功催化乙烯原位共聚得到含C6及以上长支链的LLDPE,支链度可达2.1(1000 C);随着聚合温度的升高,催化活性和支化度提高,而分子量分布则变窄,与对应的两种独立单核配合物组成的混合催化体系相比,所得聚合物支链度更高,分子量分布更窄.钛-铬异双核配合物21催化乙烯原位共聚所得的线型低密度聚乙烯以丁基支链为主.当CGC-Ti和SNS-Cr中两个金属中心之间的桥联碳原子数量减少,两个活性中心相互靠近,双金属协同效应和电子效应的影响显著提高,催化所得共聚物分子量是两种单核配合物组成的混合催化体系的18倍,支链度是其4倍.但异核双功能催化剂合成困难,并且难以保证配位金属按照1∶1的摩尔比成功配位,因此研究难度较大.

图14 烷基桥联的异核双功能催化剂20和21Fig.14 Bifunctional catalysts based on alkyl-bridged heterobimetallic complexes 20 and 21
1.3 非茂金属作共聚催化剂
与茂金属催化剂相比,非茂类过渡金属催化剂可以通过设计其结构来有效调控催化性能,因此,越来越多双功能催化体系的研究开始选用非茂金属催化剂.
YANG等[38]以锆配合物22为单一催化剂(图15),在混合助催化剂[AlEt2Cl/MAO、AlEt2Cl/AlEt3、AlEt2Cl/Al(i-Bu)3]作用下能催化乙烯原位共聚,所得聚乙烯含有多种支链结构及分布.在助催化剂AlEt2Cl的作用下,锆催化剂发挥齐聚效果,得到以1-丁烯、1-己烯和1-辛烯为主的α-烯烃,选择性可达80%,聚合物支链度最高达4.3(1000 C).当AlEt2Cl用量增大,催化活性和分子量明显下降,表现出负“共单体效应”.
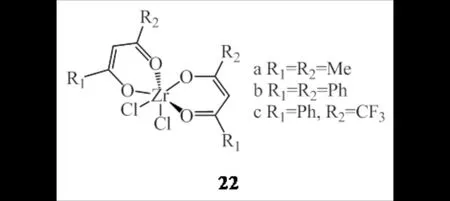
图15 β-二酮锆配合物22Fig.15 β-Diketonate zirconium complexes 22
GAGIEVA等[39]将具有齐聚性能的镍配合物23和具有共聚性能的钛配合物24组合(图16),在混合助催化剂Et2AlCl/Bu2Mg的作用下,催化所得的线型低密度聚乙烯含乙基和丁基支链,改变聚合条件可使共聚单体插入率在2%~6%范围内有效调控;叔丁基取代的配合物24a因其较小的空间位阻有利于α-烯烃的插入,因此其共聚能力大于金刚烷基取代的配合物24b.
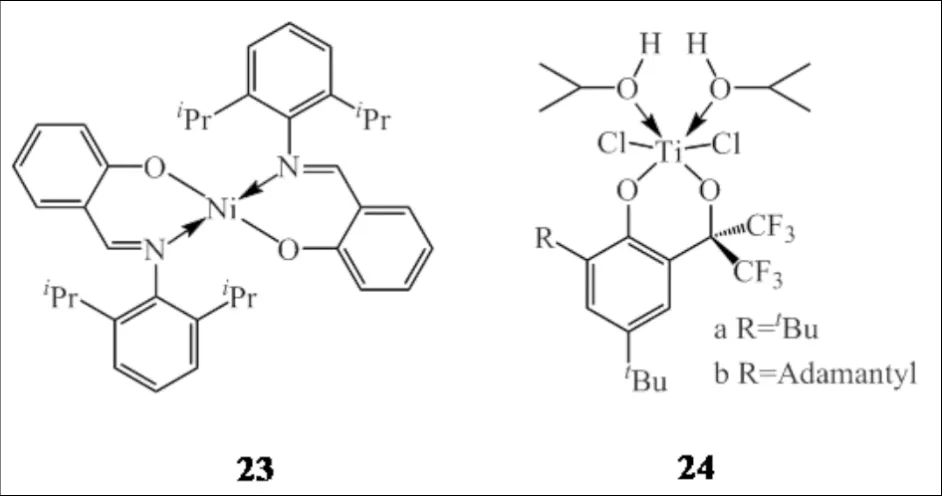
图16 镍配合物23和钛配合物24组成双功能催化剂Fig.16 Bifunctional catalysts based on nickel and titanium complexes 23 and 24
GUO等[8,40]报道了两种具有良好乙烯/α-烯烃共聚性能的新型FI-Ti配合物25(图17),并与高活性高选择性的乙烯三聚催化剂SNS-Cr(2g)组成双功能催化剂体系,在50 ℃下成功催化乙烯原位聚合制备出超高分子量的含丁基支链LLDPE,共聚物热稳定性好、分子量分布窄,并且聚合时间延长可进一步提高共聚物的分子量.
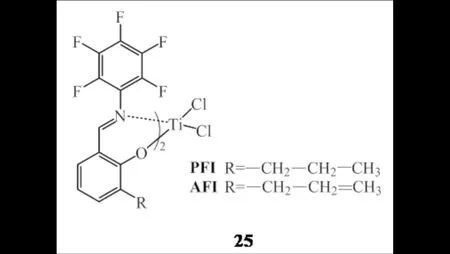
图17 FI-Ti配合物25Fig.17 FI-Ti complexes 25
XIE等[41-42]以吡啶二亚胺铁配合物(1f)分别与两种具有高乙烯/α-烯烃共聚活性的非茂金属钛配合物(26,27)组成乙烯原位共聚催化体系(图18),乙烯为唯一单体原料,MMAO为唯一助催化剂,成功催化乙烯原位共聚制备LLDPE,该聚合物含有乙基、丁基及更长支链,并且通过简单地调节反应温度等聚合条件,支化度可以得到有效地调控,最高可达39.3(1000 C).两种双功能催化体系匹配度均较高,共聚催化剂显示出强的正共单体效应.随后他们以水杨醛亚胺型双核钛配合物(28)取代上述单核钛配合物作双功能催化剂体系中的共聚催化剂[43],也能够使乙烯原位共聚得到以乙基和丁基支链为主的LLDPE,相较于对应的单核钛配合物,所得聚合物的支链度和分子量都明显提高.

图18 具有共聚活性的钛配合物26~28Fig.18 Titanium complexes 26-28 with copolymerization activity
2 结语
双功能催化剂催化乙烯原位共聚是制备线性低密度聚乙烯的重要途径,是金属有机化学和烯烃聚合研究领域的热点之一.本文综述了双功能催化剂体系催化乙烯原位共聚制备LLDPE的研究进展.按双功能催化剂体系的共聚催化剂分类,分别从Ziegler-Natta催化剂/齐聚催化剂、茂金属催化剂/齐聚催化剂和非茂金属催化剂/齐聚催化剂三种体系展开讨论;同时,原位共聚法对双功能催化剂的匹配性和反应速率搭配具有很高要求.探索合成简便高效、催化性能优异、匹配性好、选择性高的双功能催化剂体系一直是人们追求的目标;通过优化催化剂组合、修饰催化剂结构、调整聚合条件等手段可有效调控聚合性能和改善聚合物性质,为合成新性能聚烯烃材料提供了可能.
茂金属作为双功能催化剂中的共聚催化剂研究较多,茂金属催化剂虽具有活性高等诸多优点,但催化剂结构相对单一,成本较高且多受专利限制;与之相比,非茂过渡金属催化剂在催化烯烃聚合和共聚方面同样表现出优异的催化性能,且配位空间更加开阔,容易通过修饰结构达到提高催化性能的优点.因此,基于非茂类过渡金属催化剂的双功能催化剂体系具有巨大的发展潜力,催化性能优异、价格低廉的双功能催化剂将在烯烃聚合理论研究和工业生产中得到更好发展.
猜你喜欢
今日农业(2021年19期)2022-01-12
农业机械学报(2021年10期)2021-11-09
家庭影院技术(2021年7期)2021-08-14
世界有色金属(2020年1期)2020-03-26
北京航空航天大学学报(2019年3期)2019-04-08
汉语世界(The World of Chinese)(2018年6期)2018-01-22
科技创新与应用(2017年20期)2017-07-15
化学教学(2016年10期)2016-11-25
食品工业科技(2014年13期)2014-12-16
中学生数理化·高二版(2008年2期)2008-10-19
