
盐酸法磷酸萃取相的洗涤和磷酸反萃取过程初探
2021-08-12 07:21张少杰唐盛伟郑东钥钟本和陈彦逍
无机盐工业 2021年8期
张少杰,唐盛伟,王 杨,郑东钥,钟本和,陈彦逍
(四川大学化学工程学院,四川成都610065)
磷酸是磷化学工业中的重要化工产品,被广泛应用于农业、食品、制药、电子工业等国民经济部门。根据其制备过程不同,磷酸主要分为热法磷酸和湿法磷酸。其中热法磷酸根据其纯度可分为工业级磷酸、食品级磷酸、试剂级磷酸以及电子级磷酸,而湿法磷酸杂质含量较高,据统计90%以上的湿法磷酸都用于磷肥生产[1]。
湿法磷酸过程主要是指利用无机强酸(硫酸、盐酸、硝酸)分解磷矿制备磷酸,相应的湿法磷酸路线又以硫酸分解磷矿结合溶剂萃取净化磷酸较为成熟,国外在20世纪60年代后对溶剂萃取净化湿法磷酸的技术进行开发,其中以色列矿业公司(IMI)、德国巴登哈姆公司(Budenheim)、比利时普雷昂公司(Prayon)、英国奥布拉威尔森公司(Albright&Wilson)以及法国罗纳-普朗克公司(Rhone-Poulenc)等形成了专利和工业化流程[2];而中国于2003年由四川大学和贵州宏福实业有限公司开发了适合中国湿法磷酸品质的净化技术,并获得了合格的净化湿法磷酸产品[3]。
目前,国内主要使用硫酸法过程生产湿法磷酸:
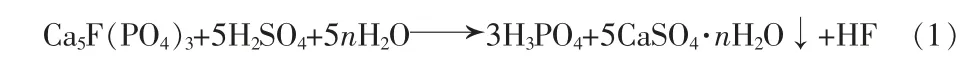
硫酸法过程中消耗硫酸的费用约占直接成本的60%[4];同时每生产1 t磷酸(P2O5计)副产4.5~5.0 t磷石膏,目前世界年磷石膏排放量为1.6亿~1.7亿t[5]。中国年排放量在2017年末已达7 500万t,对磷石膏综合利用率的要求在“十二五”之后已经提高到30%以上,但由于磷石膏中有害杂质含量高、开发利用投资较大、后沿产品附加值低、利润微薄,导致了不少企业磷石膏利用装置开工率低,存在“以销定产”的情况,大量磷石膏仍以堆存为主,占用土地资源的同时更导致环保安全出现隐患[6-7]。
中国磷矿质量较差,中低品位矿多,富矿少,全国磷矿平均品位在17%(P2O5)左右,其中富矿占磷矿总量约为8.5%[8]。而根据HG/T 2673—1995《酸法加工磷矿石》和HG/T 2674—1995《黄磷用磷矿石》,相应的湿法和热法磷酸用磷矿品位均需达到24%(P2O5)以上。综上,开发出低能耗、低成本、环境友好并且适用于中国中低品位磷矿的磷酸生产工艺尤为关键。
窑法磷酸工艺和盐酸法湿法磷酸工艺在中低品位磷矿利用方面具有其独到的优势。基于与热法磷酸相同的工艺原理,中国目前研发的窑法磷酸工艺可以利用中低品位硅质磷矿制备出纯度与热法酸相当的磷酸,且具有更好的经济效益,但由于技术原因中国的窑法磷酸仍停留在工业试生产阶段[9-10]。
而盐酸法湿法磷酸的优势在于可利用工业副产盐酸(质量分数为20%~35%)对高硅中低品位磷矿进行分解,分解速率快,磷收率高,其主化学反应式:
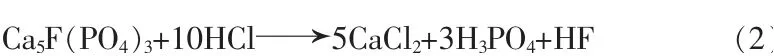
针对盐酸法工艺,以色列矿业公司(IMI)开发了相应的专利技术,实现了磷酸与CaCl2的分离,并推行工业化,用于生产饲料级和食品级磷酸。但由于IMI流程使用的混合醇类萃取剂(体积分数为60%正戊醇+40%异戊醇),溶剂回收的工序更增加了流程的复杂程度与能耗[11]。
磷酸三丁酯(TBP)被认为是一种优良的磷酸萃取剂,其具有性质稳定、水中溶解度小、对磷酸萃取效力强、对杂质净化程度高等优点。四川大学基于TBP开发出了免溶剂回收的湿法磷酸萃取净化工艺,每1 t P2O5溶剂消耗量仅为6 kg,具有良好的经济效益和工艺可行性[3]。李军等则对TBP在盐酸法体系中萃取净化湿法磷酸进行了研究,结果表明磷酸在TBP相中的分配系数随着水相中CaCl2的浓度提高而提高,TBP在盐酸法流程中具备良好的应用前景[12-13],并基于此提出了以TBP作为主要磷酸萃取剂,通过盐酸分解中低品位磷矿制备工业级和食品级磷酸的方法[14]。
盐酸法磷酸工艺的关键在于通过液-液萃取过程分离磷矿浸出液中的H3PO4和CaCl2,由于萃取H3PO4时少量CaCl2被同时萃取,需要综合考虑过程中H3PO4萃取、有机相洗涤、H3PO4反萃取3个环节,才可能最大限度地降低反萃酸中的CaCl2含量,再经过后处理使净化磷酸达到符合GB/T 2019—2008《工业磷酸》规定的氯含量标准。当以TBP萃取净化盐酸法工艺得到粗磷酸时,由于萃取有机相的黏度较大,以小相比的磷酸或水相逆流洗涤有机相去除CaCl2的操作存在困难,故采取了以一定浓度的H2SO4化学洗涤有机相去除CaCl2的方法。目前,国内外关于溶剂萃取净化湿法磷酸的研究多集中于萃取剂选取及萃取过程等方面,而针对有机相的洗涤与反萃取过程少见详细报道。笔者主要针对以TBP萃取盐酸法磷酸所得有机相的洗涤和磷酸反萃取过程进行初步探究,重点讨论不同洗涤、反萃方法和条件对反萃酸品质以及硫酸钙的结晶过程的影响,从而为开发以TBP为主要萃取剂的盐酸法磷酸净化工艺提供一定的指导。
1 实验部分
1.1 主要实验试剂
表1 为主要实验药品和试剂。

表1 主要实验药品和试剂Table 1 Main experimental drugs and reagents
1.2 实验所用磷矿及其相关分析
实验所用磷矿产自四川会东某地,其组成按照GB/T 1868、1870~1881—1995《磷矿石与磷精矿化学分析方法》系列国家标准进行测定[15],结果见表2;实验所用磷矿-盐酸浸出液为上述磷矿与质量分数为20%HCl按照理论量反应制得,其中H3PO4与CaCl2分别按照上述国标通过磷钼酸喹啉重量法以及EDTA容量法测定,其余元素含量由离子发射光谱法(ICP-OES)进行测定,其组成见表3;实验所用待反萃有机相为萃取剂(TBP与磺化煤油按照体积比4∶1均相混合)与磷矿浸出液(除铁)达到萃取平衡所得,其组成见表4。

表2 实验用磷矿组成(干基)Table 2 Composition of the experimental phosphate rock powder(dry basis) %

表3 盐酸法磷矿浸出液组成(萃取除铁后)Table 3 Composition of the phosphate ore-hydrochloric acid leach liquor(after removing Fe3+)

表4 待洗涤-反萃有机相的组成Table 4 Composition of the organic phase to be scrubbed and stripped %
1.3 主要试验仪器
DX-2700 X射线衍射仪;JSM-7500F扫描电子显微镜—能谱仪;ICP-OES 5100 SVDV电感耦合等离子发射光谱仪;Particle Track G400聚焦光束反射测量仪;FZ10蠕动泵。
2 实验结果与讨论
2.1 一步法反萃取H3PO4实验
通常,从磷矿-盐酸浸出液中萃取H3PO4时,较高的CaCl2浓度会增大H3PO4在有机相中的分配系数,促使H3PO4被萃取进入有机相,而在用纯水反萃有机相中的H3PO4时,CaCl2的浓度降低,又有利于H3PO4从有机相中萃出进入水相,提高H3PO4的回收率。本部分实验使用一定体积的硫酸溶液同时作为CaCl2沉淀剂和H3PO4反萃取剂,在反萃H3PO4的过程中同时与CaCl2发生反应,过程中产生的HCl可以在后续盐酸解析以及磷酸浓缩的过程中基本除净:
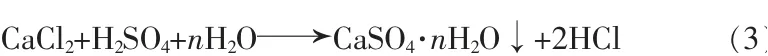
实验过程:取一定体积和质量的待反萃有机相,根据CaCl2和H2SO4的理论反应量,按照一定反萃取相比V(A)/V(O)(反萃取水相与待反萃有机相的体积比)配制相应浓度的稀硫酸,一次混合后在搅拌釜中进行恒温反萃取。在CaSO4-H3PO4-H2O三元系统中,由于半水硫酸钙(CaSO4·0.5H2O)形成晶核所需活化能最小,所以硫酸钙首先是以非稳态的半水物生成的,而在较低的磷酸浓度下,半水物将转化为二水物(CaSO4·2H2O)和无水物(CaSO4)两种稳 定晶型[8,16]。考 虑 到式(3)的 反 应 程 度 和 硫 酸 钙结晶的转化动力学,反萃实验在400 r/min下进行70 min,静置后分去上层有机相,再通过离心和抽滤分离反萃酸中的石膏固相。石膏在45℃下干燥36 h后用作SEM和XRD分析。反萃酸中的H3PO4含量以磷钼酸喹啉重量法测定,少量Ca2+通过离子发射光谱方法(ICP-OES)测定,最终H3PO4的反萃取率(SE)按照下式计算:
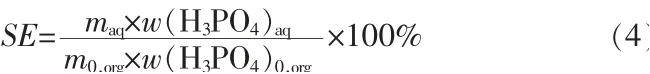
式中,maq、m0,org为反萃后水相和待反萃有机相的质量,g;w(H3PO4)aq、w(H3PO4)0,org为反萃后水相和待反萃有机相中H3PO4的质量分数,实验结果见表5。由表5可知,当使用理论量的H2SO4对有机相进行反萃时,H3PO4的反萃取率随着反萃取剂用量增加而升高,等体积[V(A)/V(O)=1∶1]反萃取时,H3PO4的反萃取率超过90%;温度升高有利于H3PO4的反萃取。相较于待反萃有机相,发现反萃水相中的磷钙比[n(H3PO4)/n(Ca2+)]显著提升,说明了硫酸作为反萃取剂对Ca2+有比较明显的去除效果,但由于反萃取磷酸中硫酸钙溶解平衡的存在,水相中的Ca2+含量始终略高于0.2%(CaCl2质量分数)。且随着反萃相比V(A)/V(O)的增大,反萃酸中H3PO4的反萃取率升高但是磷钙比n(H3PO4)/n(Ca2+)下降,这同样是硫酸钙溶解程度增大所致。

表5 不同条件下以H2SO4溶液直接反萃H3PO4的实验结果Table 5 Experimental results of H3PO4 stripping when H2SO4 solution was used as stripping agent under different conditions
图1 为不同实验组所得石膏的SEM照片。由图1可见,在理论H2SO4用量下,当体系中初始H2SO4浓度降低时,所得石膏的结晶尺寸变大,形貌逐渐向板状和长条状过度,但在搅拌作用下出现细小碎晶,所得石膏的规整度变差,该现象不利于实际生产中石膏过滤的进行。其原因是SO42-浓度下降导致了水相中硫酸钙结晶过饱和度降低,限制了结晶过程初期的成核数量,从而使晶体的几何尺寸趋向于增大,在搅拌下机械碰撞效应也更为明显,导致了晶体二次成核的发生和细晶的产生[17]。因此,通过控制硫酸浓度来控制结晶过饱和度,通过控制搅拌强度来减少结晶过程中的二次成核,对得到粗大均匀、易于过滤分离的硫酸钙结晶尤为重要。

图1 反萃取过程中生成CaSO4·nH2O的SEM照片Fig.1 SEM images of the prepared CaSO4·nH2O during extraction process
图2 为实验所得石膏的XRD谱图。由图2可以看出,所得石膏一般为无水、半水和二水的混合物。由CaSO4-H3PO4-H2O体系平衡图可知,在较高温度时(>40℃),硫酸钙晶型的转化顺序为半水→二水→无水,温度较低时(<40℃),转化顺序为半水→无水→二水[16]。实验结果表明,随着反应温度的提高,无水硫酸钙的衍射峰更加明显。同时,随着反萃硫酸初始浓度的减小,相同温度和时间条件下,半水硫酸钙的转化程度明显提高,在实验组a3条件下,半水硫酸钙已经基本完全转化。
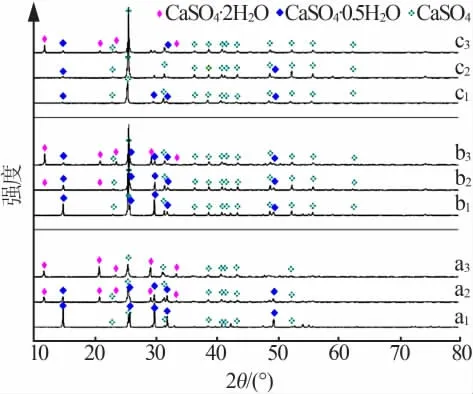
图2 反萃取过程中生成CaSO4·nH2O的XRD谱图Fig.2 XRD pattern of the prepared CaSO4·nH2O during extractin process
通过上述实验结果可知:1)在用理论量的H2SO4对有机相中的H3PO4进行反萃时,提升反萃相比V(A)/V(O)、升高反萃取温度可以提升H3PO4的反萃取率;2)降低反萃H2SO4的初始浓度可以得到几何尺寸更大的硫酸钙结晶,但此时晶体的破碎和二次成核现象也更为明显,导致大量碎晶的产生,因此需要对反萃硫酸浓度和搅拌强度加以控制来得到粗大均匀、易于过滤的硫酸钙结晶;3)由于受到硫酸钙溶解化学平衡的限制,通过H2SO4溶液直接反萃有机相中的H3PO4无法进一步提高H3PO4和CaCl2的分离程度,因此下文中提出了CaCl2洗涤-H3PO4反萃的二步法流程,即先用少量H2SO4溶液洗涤有机相,分离掉硫酸钙石膏后,再以软水反萃取有机相中H3PO4,从而进一步降低Ca2+在反萃磷酸中的含量。
2.2 洗涤-反萃两步法实验
2.2.1 洗涤实验
在使用理论量H2SO4对有机相进行洗涤去除CaCl2的过程中,为了降低被洗水带走的H3PO4损失,采用了小相比[V(A)/V(O)=1∶10]进行洗涤。该过程较为复杂,涉及到硫酸液滴在有机相中的分散、H2SO4与CaCl2在液滴和有机相界面间的传质和反应、CaSO4·nH2O晶体的非均相成核以及生长等过程,因而研究难度较大。
聚焦光反射测量技术(FBRM)可以用来研究分散在液体中的液滴或者微粒,获取其尺度和数量的信息。图3[18]为FBRM的工作原理,在研究液体中的结晶过程时,聚焦光束运动方向上扫过晶体微粒的距离被记录为微粒的弦长(chord length),不同弦长区间内微粒的计数每个检测扫描周期内检测到微粒的个数为弦长分布(CLD),弦长分布可以一定程度上反映应晶体生成过程中的晶体尺度分布(CSD)变化。实验中采用聚焦光束反射测量仪对洗涤过程进行研究,对有机相中硫酸钙晶体的生成规律进行在线观测。实验原理见图4,实验温度为20℃,搅拌转速为400 r/min,调节蠕动泵转速使洗涤硫酸在1 min时完成进料,进料开始同时以聚焦光束反射测量仪记录结晶过程数据。
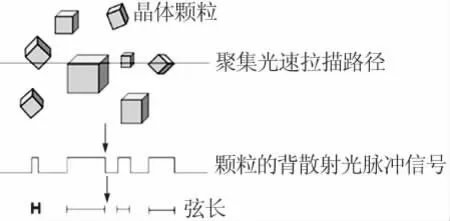
图3 聚焦光反射测量仪检测结晶过程原理图[18]Fig.3 Crystallizing process observed by FBRM[18]
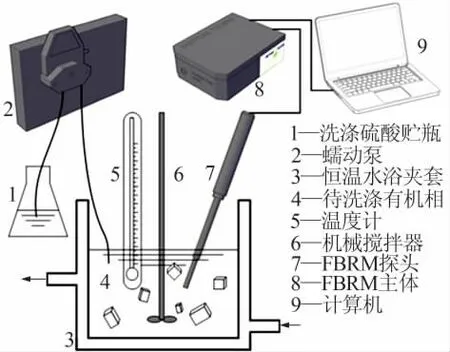
图4 洗涤过程实验原理图Fig.4 Experimental schematic diagram of the scrubbing process
由于FBRM方法可以同时检测到分散在有机相中的水相微液滴和洗涤过程中生成的硫酸钙结晶。故洗涤试验前先进行了空白实验以确定液滴分散过程对结晶检测的影响,即在相同的实验条件下测试了硫酸在未萃取H3PO4和CaCl2的空白有机相(萃取剂组成80%TBP+20%煤油,体积分数)中的分散情况,相比为V(A)/V(O)=1∶10,结果见图5。图5表明,洗涤酸在空白有机相中主要形成了弦长<10 μm和10~50 μm的微液滴,二者计数和变化趋势基本相当,均随搅拌时间的增加而增加,30 min时趋于平稳;空白实验中基本未见大液滴的生成,说明了在搅拌转速和加料相比下洗涤硫酸在有机相中的分散良好。
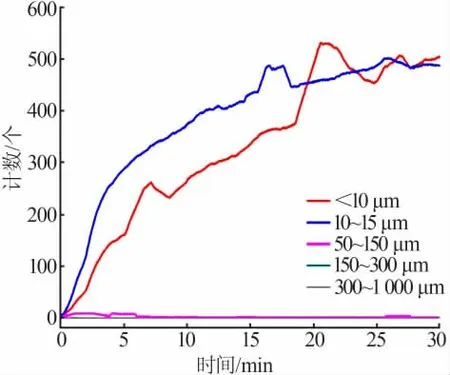
图5 反萃硫酸水溶液在有机相中的分散实验(空白实验)Fig.5 Dispersion experiment of sulfuric acid droplets in organic phase(blank experiment)
图6 为洗涤过程中硫酸钙晶体生成的规律。由图6可知,在开始匀速进料洗涤硫酸时,有机相中立刻出现大量弦长<10 μm和10~50 μm的微粒,其计数远高于图5中洗涤硫酸在空白实验中有机相中分散成微液滴时的计数,说明此时有机相中生成的结晶微粒数量占主导地位,液滴的分散对结晶过程的检测没有明显影响;同时可以看出,开始加料时体系中小微粒的计数增长得非常快,这说明了洗涤硫酸液体和有机相的界面处存在相当大的过饱和度和较强的搅拌扰动,使得硫酸钙晶核迅速大量形成,在洗涤过程中晶体的成核诱导期非常短,直接进入成核阶段;同时一开始形成的少量较大微粒(弦长50~1 000 μm)的消失,说明了在结晶过程中有局部微粒的破碎和溶解;在0~8 min硫酸钙晶体的成核和生长基本完成,此后基本进入体系中残留过饱和度消耗阶段,此时体系过饱和度已经大大降低,不再生成新的晶核,弦长<10 μm和10~50 μm的微粒计数部分下降,较大颗粒(>50 μm)的计数开始上升,同时实验观察到有机相的透明度增加,部分微粒聚并成为较大的无法检测的结块聚沉。图7为洗涤过程中不同时刻弦长分布(CLD)。由图7可知,随着洗涤时间的增加,微粒的计数和弦长分布呈现了由小到大再逐渐变小的规律,说明了硫酸钙晶体的形成基本遵循成核、生长、体系中过饱和度消耗和结块沉降的过程。

图6 洗涤过程中微粒计数变化Fig.6 Counts of particles vary with time in the scrubbing process

图7 洗涤过程中微粒弦长分布(CLD)—时间变化Fig.7 Chord length distribution(CLD)varies with time in the scrubbing process
图8 和图9分别为洗涤时间为10 min时,上述过程所得洗涤石膏的SEM照片与XRD谱图。由图8可以看出,洗涤石膏主要由最大长度为20 μm的硫酸钙晶体搭接构成;由图9可见,洗涤10 min时,所得石膏的物相组成为CaSO4·0.5H2O。

图8 洗涤硫酸钙的SEM照片Fig.8 SEM images of CaSO4·nH2O in the scrubbing process
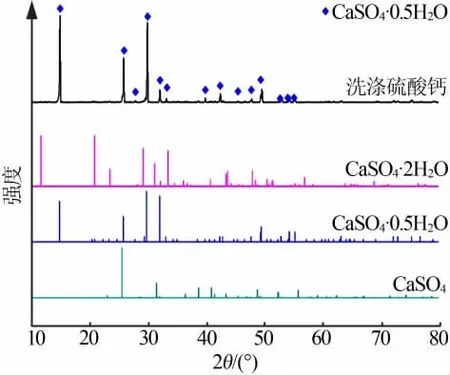
图9 洗涤硫酸钙的X射线衍射谱图Fig.9 XRD pattern of CaSO4·nH2O in the scrubbing process
2.2.2 洗涤有机相磷酸反萃取实验结果
对上述洗涤条件下所得有机相用去离子水进行反萃,仍按照式(4)对整个洗涤-反萃过程中磷酸的反萃取率进行计算,结果见表6。
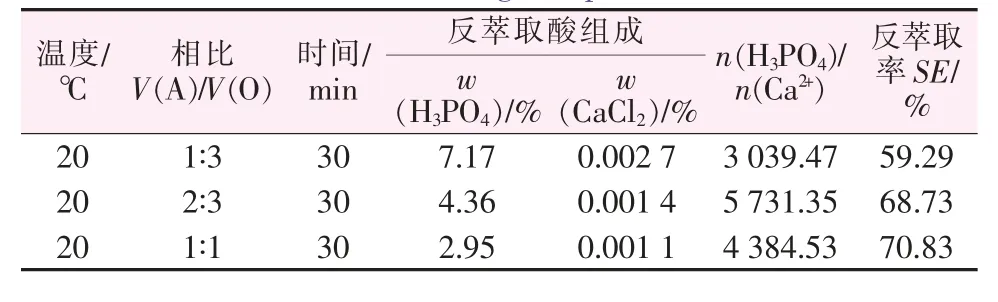
表6 洗涤-反萃过程实验结果Table 6 Results of H3PO4 stripping from the scrubbed organic phase
表6 结果说明,在20℃时对洗涤后的有机相进行反萃,当反萃取相比V(A)/V(O)为1∶3、2∶3和1∶1时,H3PO4的反萃取率分别为59.29%、68.73%和70.83%。而对比表中5中a1~a3组的实验结果,发现在相同的反萃取温度和相比V(A)/V(O)下,采用H2SO4溶液对有机相直接进行反萃取时,H3PO4的反萃取率分别为56.33%、85.92%、92.28%。显然,在较高的反萃取相比[V(A)/V(O)为1∶1]时,采用洗涤-反萃流程,H3PO4的反萃取率降低了20%左右,这部分H3PO4反萃取率的损失应该为洗涤过程中有机相中的部分H3PO4进入洗涤水相被带走所致,而实际流程中可将洗水返回萃取工段以回收其中的H3PO4;而从磷钙比[n(H3PO4)/n(Ca2+)]来看,洗涤反萃酸中的磷钙比均大于3 000,且随着反萃取水相用量的增大,磷钙比并没有减小,这是因为分离洗涤石膏固相后消除了CaSO4·nH2O溶解平衡对反萃酸品质的影响。对比表5中a1~a3组的实验结果发现,通过洗涤-反萃流程得到的反萃酸,其磷钙比增大了65倍以上,与直接采用H2SO4溶液对有机相中的H3PO4进行反萃相比,采用先以H2SO4进行洗涤有机相分离硫酸钙石膏后再以软水反萃取H3PO4的流程,可显著降低反萃取磷酸中Ca2+含量。
2.3 洗涤石膏的综合利用分析
常见的硫酸法湿法磷酸生产工艺按照其所得的水合硫酸钙结晶类型分为二水物法、半水物法和再结晶法(半水-二水法和二水-半水法)。以本实验用磷矿(表2)为例,采用二水法生产工艺(80℃,搅拌转速为400 r/min,搅拌时间为4.5 h,反应料浆液固质量比为3∶1,制备磷酸质量分数为21.32%(P2O5),磷矿粉过0.178 mm筛),实验室制备的磷石膏SEM、EDS、XRD图分别见图10、11。
图10 表明,以二水物法工艺处理本实验所用磷矿所得磷石膏的主要物相成分为CaSO4·2H2O和SiO2;图11表明,硫酸钙固相为典型的斜方六面板状和棱柱状晶体,面扫图像表明磷石膏中混合着较大量的不规则的酸不溶SiO2。而SiO2作为硫酸法石膏中的主要固体杂质,对磷石膏的综合利用会产生不利的影响,比如在磷石膏制硫酸联产水泥工艺中,磷石膏中SiO2含量对磷石膏热分解生产工艺和产品质量影响较大,采用高硅含量的磷石膏时热分解过程中易结圈,水泥熟料产品质量不稳定;在磷石膏制备硫酸铵和碳酸钙的过程中,磷石膏中的高SiO2含量会导致其副产的碳酸钙产品质量不合格,不能作为市场产品销售从而降低了磷石膏综合利用的可行性,增大了磷石膏处理的难度等[19-20]。本文的研究结果表明,采用盐酸法工艺流程时得到的石膏属于清洁白石膏,不含SiO2等固体杂质,这就很大程度上降低了工艺中石膏的处理难度,提升了其综合利用价值和潜力。
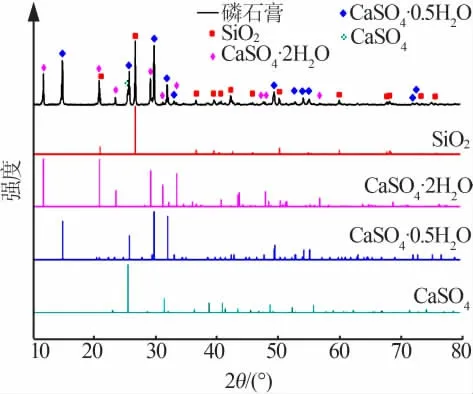
图10 四川会东磷矿磷石膏的X射线衍射谱图Fig.10 XRD pattern of phosphogypsum generated by phosphate rock in Huidong,Sichuan

图11 四川会东磷矿磷石膏的扫描电镜/能谱面扫图像Fig.11 SEM and EDS mapping images of phosphogypsum generated by phosphate rock in Huidong,Sichuan
从产生石膏的量来看,以传统二水硫酸法工艺为例,100份质量的磷矿完全被H2SO4分解时的石膏值可按照下式计算:

式中:w(CaO)为磷矿中CaO质量分数,%;w(AI)为磷矿中酸不溶物质量分数,%;172.1为CaSO4·2H2O摩尔质量,g/mol;56为CaO摩尔质量,g/mol。
对于本实验中所用磷矿(表2),若以SiO2含量近似代表总酸不溶物含量AI,其100份质量磷矿产生的干基磷石膏计算值约为137份,其中主要成分为CaSO4·2H2O和SiO2。如按照本文中的盐酸法工艺,假设被萃取入有机相中的CaCl2全部以CaSO4·2H2O的形式成为洗涤石膏,则此工艺中的理论石膏值:
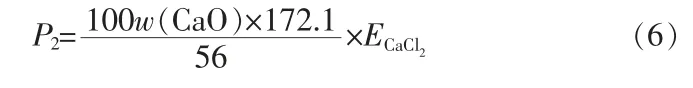
式中:ECaCl2为萃取过程中CaCl2的萃取率。
之前的实验工作表明,通过萃取剂(80%TBP+20%煤油,体积分数)逆流萃取上述磷矿-盐酸浸出液中的H3PO4,经四级逆流萃取后CaCl2的共萃取率在10.27%左右,按照此值计算所得的洗涤石膏值P2约为11.9份,仅为二水硫酸法理论石膏值的8.7%左右。可见在工业副产盐酸来源有保障、CaCl2盐水后处理技术相对成熟的情况下,盐酸法路线在节约硫资源消耗和减少磷石膏排放方面可以展现出其巨大的优势。
3 结论
1)采用H2SO4溶液一步沉淀有机相中的CaCl2并反萃取H3PO4时,提高反萃取剂的体积用量和反萃取温度可增大H3PO4的反萃取率;60℃时,当反萃取的相比V(A)/V(O)=1∶3时,H3PO4反萃取率可达75%以上,相比V(A)/V(O)增大到1∶1时,H3PO4反萃取率可达96%以上。硫酸钙沉淀的晶体尺寸主要受到H2SO4浓度的影响,H2SO4浓度降低,硫酸钙晶体向长条板状过度,其长径比增大明显,同时会出现细小碎晶,过低的H2SO4浓度不利于硫酸钙的结晶和过滤质量的提高。由于硫酸钙在反萃酸中存在溶解平衡,因此反萃磷酸中的CaCl2质量分数始终略高于0.2%,此法对反萃磷酸的净化效果有限。
2)洗涤反萃两步法流程实验的结果表明,在20℃,相比V(A)/V(O)分别为1∶3、2∶3和1∶1,以软水对洗涤后有机相中的H3PO4进行反萃取,H3PO4的反萃取率分别为59.29%、68.73%和70.83%,有机相中H3PO4的洗涤损失率为20%左右,但此时反萃酸中磷钙比n(H3PO4)/n(Ca2+)均>3 000,约为用H2SO4直接反萃时的65倍以上,反萃酸中Ca2+的去除率显著提高。聚焦光反射测量技术对洗涤过程的检测表明,实验条件下H2SO4液滴可在有机相中良好分散,洗涤过程在10 min内基本完成,生成的石膏均为CaSO4·0.5H2O。
3)本实验中所得硫酸钙均为不含SiO2的清洁石膏,与硫酸法工艺中产生的二水石膏相比,其后处理难度小,综合利用潜力大,且其石膏产量仅为二水硫酸法理论石膏产量的8.7%左右,显著降低了湿法磷酸过程中的石膏处理量。
猜你喜欢
有色金属(矿山部分)(2021年4期)2021-08-30
矿产勘查(2020年5期)2020-12-19
中华养生保健(2020年3期)2020-11-16
盐科学与化工(2020年3期)2020-04-26
活力(2019年21期)2019-04-01
中国化肥信息(2018年5期)2018-08-04
中国资源综合利用(2017年4期)2018-01-22
中国民族医药杂志(2016年9期)2016-05-09
人间(2015年11期)2016-01-09
中国塑料(2015年1期)2015-10-14
