
提取方法对马铃薯渣果胶多糖组成及分子链构象的影响
2021-08-11 09:33孙玮璇田金虎陈健乐陈士国刘东红叶兴乾
中国食品学报 2021年7期
孙玮璇,田金虎,陈健乐,陈士国,刘东红,叶兴乾*
(1浙江大学生物系统工程与食品科学学院 馥莉食品研究院 浙江省农产品加工技术研究重点实验室智能食品加工技术与装备国家地方联合工程实验室 浙江省食品加工技术与装备工程实验室 杭州 310058 2浙江大学宁波研究院 浙江宁波 315100)
薯渣是马铃薯淀粉加工中产生的废弃物,每生产1 t 马铃薯淀粉约产生0.75 t 的薯渣[1]。目前主要有2 种马铃薯渣处理途径:一种是用作动物饲料[2],操作简单且技术含量低,然而,薯渣中粗纤维含量较高,蛋白质含量偏低,导致薯渣饲料适口性较差,热量较低,且存在易腐败,脱水能耗高等问题,制约了薯渣饲料的开发[3]。另一种是将薯渣直接废弃、露天堆放或做掩埋处理,该方法不仅造成资源的极大浪费,还带来严重的环境污染[3]。有学者研究发现马铃薯渣中含有丰富的细胞壁多糖(70%),其中果胶占主要部分(56%)[2],因此从马铃薯渣中提取果胶多糖,成为一种潜在的提高马铃薯渣附加值的有效手段。
传统果胶多糖(柑橘、苹果来源)以均聚半乳糖醛酸聚糖结构(HG)为主,主要由α-(1,4)链接的半乳糖醛酸(GalA)构成。依据GalA 残基中甲酯化修饰程度不同,果胶多糖可分为高甲氧基果胶(HMP,DM>50%) 和低甲氧基果胶(LMP,DM<50%)[4]。不同于传统柑橘来源果胶多糖,马铃薯果胶多糖中HG 结构占比较少,主要为鼠李半乳糖醛酸聚糖I(RG-I)结构(75%)[2],RG-I 主链以半乳糖醛酸(GalA)和鼠李糖(Rha)由α-(1,4)和α-(1,2)糖苷键交替链接而成,主链上20%~80%的鼠李糖残基在其O-4 处存在中性糖链侧链分支(半乳聚糖、阿拉伯聚糖、阿拉伯半乳聚糖)[5]。一般认为,较少的HG 结构和高度分支的RG-I 结构使得马铃薯果胶多糖的凝胶能力较弱,这限制了马铃薯果胶的商业应用[6]。然而,近年来研究人员发现富含RG-I 结构的果胶多糖具有较高的健康功能活性(抗癌,抗增生,肠道益生性等)[7],因此,富含RG-I 结构的马铃薯果胶多糖作为一种功能性多糖而备受关注。RG-I 型果胶多糖的肠道益生活性主要归因于其半乳糖含量丰富[8]。体外研究发现,果胶多糖末端β-半乳糖含量越多,肠道益生菌生长速率越高[9]。另外,RG-I 型果胶多糖降解后的低分子质量多糖和寡聚糖比原果胶更能促进肠道双歧杆菌的生长[10-11]。通常认为RG-I 型果胶多糖的抗癌、抗增生等生理活性与其β-半乳糖和半乳糖凝集素-3 的结合有关[12],而也有研究证明保留适当的HG 片段会通过其它机理抑制癌细胞增殖[13]。化学组成和分子质量决定果胶多糖的功能活性,此外,酯化取代等修饰也可能影响某些保健功效的发挥[14-16]。
由于果胶多糖的结构易受提取条件的影响,因此了解不同提取方式下果胶多糖结构组成差异,就能从提取开始有针对性地筛选提取条件,从而直接或经简单改性得到特定活性较高的果胶多糖组分。碱法和酶法常用来提取富含RG-I 结构域的果胶多糖。在碱法提取中,果胶多糖的HG 结构会通过β-消除作用被水解,使得富含中性糖的RG-I 结构占比更高[17]。而在酶法提取中,多聚半乳糖醛酸水解酶可以酶解GalA 残基之间的α-(1,4)糖苷键,从而降解HG 结构[18],并在较为温和的条件下制备富含RG-I 结构域的果胶多糖。不同于上述两种方法的酸法提取会造成RG-I 上中性糖侧链的降解,而HG 结构中GalA 上的酯化基团则会得到较好的保留[4]。工业上,为了保证商品果胶的凝胶特性,也多选择酸性条件提取。
目前酸法、碱法和酶法提取马铃薯果胶多糖的研究虽然较多[2,4,18],但是有关不同提取方式制备的果胶多糖在结构及性质上的差异还未见报道。本文以马铃薯渣为原料,采用酸提、碱提、酶提3 种方法提取马铃薯渣果胶多糖,比较不同提取方法的果胶得率及单糖组成差异,通过傅里叶红外光谱(FTIR)和核磁共振氢谱(1HNMR)判断果胶多糖的甲酯化和乙酰化程度,采用激光光散射法测定马铃薯渣果胶多糖的分子质量以及链形态。以期为后续马铃薯渣的资源化利用提供理论依据。
1 材料与方法
1.1 材料与试剂
马铃薯渣,宁夏佳利源薯业有限公司。耐高温α-淀粉酶(来自黑曲霉Aspergillus niger,酶活力≥2 100 U/g)、淀粉转葡糖苷酶(来自黑曲霉,酶活力≥260 U/mL)、多聚半乳糖醛酸水解酶(来自黑曲霉,酶活力为2.5 U/μL),美国Sigma-Aldrich有限公司;K-TSTA 总淀粉检测试剂盒,爱尔兰Megazyme 公司;单糖标准品:甘露糖(Man)、岩藻糖(Fuc)、鼠李糖(Rha)、阿拉伯糖(Ara)、半乳糖(Gal)、木糖(Xyl)、葡萄糖(Glc)、半乳糖醛酸(GalA),上海阿拉丁生化科技股份有限公司。其它试剂均为分析纯级,国药集团化学试剂有限公司。
1.2 仪器与设备
Waters e2695 高效液相色谱(HPLC)、2489 UV/Vis 检测器,美国 Waters 公司;DAWN HELEOS II 多角度激光光散射仪(MALLS)、RID-20A 示差检测器、LC-20 高效液相色谱仪、UV-2600 紫外可见分光光度计,日本Shimadzu 仪器有限公司;SB-805 HQ 凝胶色谱柱、SB-806 HQ 凝胶色谱柱,日本Shodex 电工科学仪器有限公司;Zorbax Eclipse XDB-C18柱,美国Agilent 科技有限公司;AVA TAR370 傅里叶红外变换光谱仪,美国Nicolet 公司;Avance Ⅲ600 MHz 核磁共振波谱仪,德国Bruker 公司。
1.3 方法
1.3.1 马铃薯渣原料中淀粉的去除 参考Khodaei 和Karboune[2]的方法。将干燥马铃薯渣粉碎过60 目筛,精确称取10 g,按照1∶15 的料液比混入磷酸盐缓冲液中(预热至95 ℃,pH 6.5),保持5 min。待温度降至85 ℃时,加入1 mL 耐高温α-淀粉酶,保持15 min。再加入300 mL 磷酸盐缓冲液(pH 6.5),待温度降至45 ℃时,加入1 mL 淀粉转葡萄糖苷酶,150 r/min 磁力搅拌下保持16 h。过滤并将滤渣用去离子水洗涤2 遍,然后将滤渣在50℃烘箱中干燥24 h。粉碎过60 目筛得到去除淀粉的马铃薯渣后,用K-TSTA 总淀粉检测试剂盒测残余淀粉含量。
1.3.2 马铃薯渣中果胶的提取
1)酸法提取 按照Yang 等[4]的方法,称取5 g 除淀粉薯渣,按照1∶15 的料液比投入去离子水中,用10%的柠檬酸,将溶液pH 值调节至2.0。80℃下250 r/min 磁力搅拌3 h,冷却至室温(25 ℃)后,用G3 砂芯漏斗过滤得到清液,加入4 倍体积的无水乙醇在4 ℃下醇沉12 h 后,6 000 r/min 离心得醇不溶物,将不溶物用无水乙醇洗涤3 次后,重新溶于去离子水中,真空冻干后得到酸法提取的马铃薯渣果胶多糖(Potato pulp pectic polysaccharide extracted by citric acid,CPP)。
2)碱法提取 称取5 g 除淀粉薯渣,按照1∶15 的料液比投入到0.5 mol/L 的氢氧化钠溶液中,80 ℃下250 r/min 磁力搅拌3 h。参考酸法提取过程中的过滤、醇沉、洗涤等操作,经冻干后得到碱法提取的马铃薯渣果胶多糖(Potato pulp pectic polysaccharide extracted by base,BPP)。
3)酶法提取 参考Øbro 等[18]的方法,称取0.5 g 除淀粉薯渣,按质量分数0.125%投入到200 mL 0.2 mol/L 的碳酸钠溶液中,4 ℃静置24 h 后,用0.1 mol/L 的醋酸钠溶液将溶液pH 值调至4,加入100 μL 多聚半乳糖醛酸水解酶,45 ℃水解20 h。经过与(1)、(2)中相同的过滤、醇沉、洗涤等步骤后,冻干得到酶法提取的马铃薯渣果胶多糖(Potato pulp pectic polysaccharide extracted by enzyme,EPP)。
1.3.3 单糖组成的测定 将上述酸、碱、酶提果胶多糖分别经衍生后,采用高效液相色谱(HPLC)测定其单糖组成[19]。具体衍生及测定方法如下:精确称取3 mg 样品于安瓿瓶中,加入2 mL 三氟乙酸(2 mol/L)使其充分溶解,110 ℃水解8 h 后氮气吹干。将吹干的水解物重新溶解于0.3 mol/L 的氢氧化钠溶液,并且加入450 μL 0.5 mol/L 的1-苯基-3-甲基-5-吡唑啉酮,在70 ℃条件下衍生30 min。衍生结束后,用0.3 mol/L 的盐酸将体系pH值调至7.0,用氯仿重复萃取3 次(每次加1 mL 氯仿)。萃取后的上层水溶液过0.22 μm 水系滤膜,并转移到进样瓶中。每个样品进样量为10 μL,25 ℃下,通过XDB-C18 柱进行分离,并由2489 UV/Vis 紫外检测器进行检测。流动相A 相为15%乙腈的磷酸二氢钾缓冲液(0.05 mol/L,pH 6.9);B相为40%乙腈的磷酸二氢钾缓冲液(0.05 mol/L,pH 6.9)。以1 mL/min 的洗脱速率进行梯度洗脱30 min,开始的10 min 内,B 相由0%线性变化到15%,之后的20 min 内由15%线性变化至25%。
1.3.4 分子质量的测定和果胶分子链构象的鉴定采用激光光散射的方法,将高效排阻色谱、激光光散射、示差检测器和粘度计联用(HPSECMALLS-RI-VISC)测出3 种马铃薯渣果胶多糖的分子质量,并且计算Mark-Houwink-Sakurada 方程以评估果胶多糖的链形态[20]。将3 mg 样品溶解于1 mL 流动相(含0.002%叠氮化钠的0.2 mol/L氯化钠),过0.22 μm 水系滤膜待进样,进样量为25 μL,串联SB-806 HQ 凝胶色谱柱和SB-804 HQ 凝胶色谱柱进行分离。25 ℃下由RID-20A 示差检测器进行检测。以0.5 mL/min 的速率洗脱50 min。折光率增量dn/dc 值为0.138 mL/g,分子质量以及其它相关数据由ASTRA 7.1.2 软件进行收集和分析。
1.3.5 甲酯化度(DM)和乙酰化度(DA)的测定根据Müller-Maatsch 等[21]的方法,采用1HNMR的图谱计算。精确称量果胶多糖样品30 mg,充分溶解在含有0.4 mol/L 氢氧化钠的重水中,室温下放置2 h 后,3 000×g 离心10 min,取上清液,加入100 μL 2 mg/mL 的三甲基硅烷基丙酸(TSP)标品(以重水为溶剂),充分混合后过0.45 μm 水系滤膜,并转移到核磁管中。采用Avance III 600 MHz核磁共振波谱仪进行扫描,每个样品扫描64 次。甲醇、乙酸和TSP 内标的信号分别为:乙酸(AcOH),1.92 ppm;甲醇(MeOH),3.36 ppm;TSP,0 ppm。甲酯化度(DM)和乙酰化度(DA)通过公式(1)~(3)计算:

式中,A——乙酸、甲醇或者TSP 在其信号处的峰面积;EW——相应物质的相对分子质量/信号内氢分子的个数;GalA%——半乳糖醛酸的质量分数,参照Blumenkrantz 和Asboe-Hansen[22]的糖醛酸测定方法,以GalA 为标准品测得。
1.3.6 傅里叶变换红外光谱(FTIR) 参考支梓鉴等[23]的方法,称取干燥的果胶多糖样品1 mg,加入40 mg 溴化钾研磨后压片,在4 000~400 cm-1波长范围,以4 cm-1的分辨率,用AVA TAR370 傅里叶红外变换光谱仪进行扫描。
1.4 数据分析
测定结果用“平均值±标准差”表示。数据由IBM SPSS statistics 23 软件进行分析,组间差异采用Tukey 事后检验确定,显著性水平为P<0.05。
2 结果与分析
2.1 马铃薯渣果胶多糖的得率以及单糖组成
除淀粉后,测得马铃薯渣中残余总淀粉含量<3.0%,采用酸提、碱提、酶提3 种方法从马铃薯渣中提取果胶多糖。不同方法下的得率如表1所示,碱提薯渣果胶多糖(BPP)得率最高为23.1%;其次为酸提薯渣果胶多糖(CPP),得率为11.7%;酶提薯渣果胶多糖(EPP)的得率最低为6.0%。单糖组成结果以摩尔比(mol%)的形式计算(表1),酸、碱、酶3 种提取方法得到的薯渣果胶多糖的单糖组成中,Gal 均占主要部分(41.0~49.6 mol%),说明马铃薯渣果胶多糖以半乳糖为主,此结果与之前报道的马铃薯渣果胶的单糖组成相符[2,4,18]。果胶多糖的HG 结构主要由GalA 构成,而RG-I结构的主链由Rha 和GalA 交替连接构成,因此Rha/GalA 的摩尔比值常用来反映RG-I 结构的多少[24]。以HG 结构为主的商品果胶Rha/GalA 值较低(0.017~0.027)[4],而以RG-I 结构为主的果胶多糖Rha/GalA 值在0.05~1 之间,比值越接近1 则果胶多糖组分中RG-I 结构占比越多,HG 结构越少[24]。CPP、BPP 和EPP 的Rha/GalA 比值在0.26~0.81 之间,表明酸、碱、酶3 种方法提取的马铃薯渣果胶多糖都以RG-I 结构为主。其中,CPP 的Rha/GalA 值最小为0.26,说明酸性条件下果胶多糖中保留了较多的HG 结构。碱法提取过程中HG结构域因β-消除作用被大量水解[2],导致HG 结构占比下降,因而BPP 的Rha/GalA 最高(0.81)。EPP 的Rha/GalA 值为0.42,低于BPP,说明酶法提取时仍有部分的HG 结构未被酶水解。Øbro 等[18]研究发现,多聚半乳糖醛酸水解酶的催化水解能力与半乳糖醛酸聚糖的聚合度相关,当HG 结构中半乳糖醛酸聚糖聚合度小于4 时,酶解能力将显著下降,由此推断EPP 中的HG 链长可能较短。

表1 不同方法提取马铃薯渣果胶多糖的得率、单糖组成、甲酯化度和乙酰化度Table 1 Yields,monosaccharide composition,DM and DA of different potato pulp pectic polysaccharides
Ara 和Gal 以中性糖侧链(阿拉伯聚糖、半乳聚糖和阿拉伯半乳聚糖) 的形式存在于果胶多糖的RG-I 结构域中,二者在EPP 中含量均高于CPP 和BPP,这说明EPP 的中性糖侧链最为丰富。因此相较于酸提和碱提,酶提取可能带来更高的分支度。CPP 中Gal 含量与BPP 无显著差异(P >0.05),而Ara 则低于BPP,这可能与阿拉伯聚糖更容易被酸降解有关[25]。
Glc 和Xyl 在3 种马铃薯渣果胶多糖中也有一定的占比,且相比于酸法和酶法,碱法提取的BPP 中二者含量更高。Glc 和Xyl 主要构成葡聚糖、木葡聚糖和木聚糖,是纤维素和半纤维素的主要成分。根据Zykwinska 等[17]的报道,马铃薯细胞壁中阿拉伯聚糖和半乳聚糖侧链易与纤维素、半纤维素发生交联,形成原果胶网络[26],碱性条件下原果胶网络会松弛[27],在提取果胶多糖的同时,纤维素、半纤维素多糖也会在碱性条件下溶出、保留,并随之提取出[2]。因此碱性条件下提取的果胶多糖中会有较多的纤维素、半纤维素组分。中性糖Fuc 和Man 也属于纤维素和半纤维素的组成成分,而在三者中含量都不多,且无显著差异(P>0.05)。
2.2 傅里叶变换红外光谱(FTIR)分析
3 种马铃薯渣果胶多糖的红外光谱如图1所示,3 410 cm-1处的宽吸收峰是由O-H 伸缩振动造成[28],2 930~2 940 cm-1处的较弱吸收峰是由CH 伸缩振动造成[29]。1 720 cm-1和1 610 cm-1处的吸收峰分别由酯化羧基和游离羧基的伸缩振动造成[29]。1 720 cm-1处峰面积与1 610 cm-1峰面积比在一定程度上反映了果胶的酯化程度[30]。CPP 在1 720 cm-1处有较强的吸收峰,而BPP 中未见吸收峰,说明碱法提取后马铃薯渣果胶多糖中基本无酯化的羧基存在。酶法提取的EPP 在1 720 cm-1处有一个较弱的吸收峰,说明EPP 中有少部分GalA 的羧基被酯化。

图1 不同马铃薯渣果胶多糖的傅里叶红外光谱图Fig.1 FTIR spectra of different potato pectic polysaccharides
1 200~900 cm-1之间是多糖的指纹区域,通常反映不同单糖组成以及它们之间的链接方式[31]。指纹区内各糖苷键的吸收峰之间存在重叠,从而导致具体的单糖组成和链接方式的差异很难通过图谱阐明。1 154~1 150 cm-1处的吸收峰是由糖苷键C-O-C 的伸缩振动造成的[32]。在指纹区波段,BPP 于1 045 cm-1处有一明显的吸收峰,而CPP和EPP 中并没有表现,此峰可能是由于木糖、葡萄糖等中性单糖的C-O 形变振动形成的[33],说明BPP 的中性糖组分中含有纤维素、半纤维素成分。
2.3 甲酯化度(DM)与乙酰化度(DA)计算
以TSP 为内标,通过1HMNR 积分峰面积计算出酸提、碱提和酶提果胶多糖的甲酯化度(DM)和乙酰化度(DA)(表1)。3 种果胶的DM 远低于50%,属于低甲氧基果胶多糖的范畴[4]。其中BPP未检出甲酯化,EPP 仅有少部分的GalA 中有甲酯化修饰,DM 为1.6%。而CPP 的甲酯化程度较高,DM 为7.5%。甲酯化主要发生在HG 结构中,碱性条件下HG 中GalA 上存在的甲氧基会因为皂化作用而水解,变成游离羧基[30]。因此,碱法提取中碱性条件和酶法提取前的弱碱条件会使得甲酯化程度大大下降。碱法提过程中的热碱环境又将进一步通过β-消除作用降解HG 结构,使得甲氧基含量进一步减少[34]。
马铃薯渣果胶GalA 上的乙酰化修饰除发生在HG 结构外,也大量的存在于RG-I 结构中[35]。3种马铃薯渣果胶的DA 均高于商品柑橘果胶多糖(1.4%~1.6%)[36]。其中,CPP 和EPP 的DA 值分别为13.6%和10.7%,均大于8%,属于高乙酰基果胶多糖的范畴[4]。BPP 的DA 值要小于CPP 和EPP,碱提过程中HG 结构的大量降解会造成乙酰基的减少[34],因而BPP 中的乙酰化修饰可能更多的存在于RG-I 结构中。
2.4 马铃薯渣果胶多糖的分子质量
酸、碱、酶3 种提取方法得到的薯渣果胶多糖的分子质量参数,包括重均分子质量(Mw)、数均分子质量(Mn)和分散度(Polydispersity,Mw/Mn),如表2所示。EPP 和BPP 分别有着最高(1 706.3 ku)和最低的Mw(520.1 ku),而CPP 的Mw 介于二者之间(752.3 ku)。结合单糖结果来看,EPP 中半乳聚糖、阿拉伯聚糖等中性糖侧链含量最为丰富,较高的分支化程度使得其拥有较高的Mw。而相比于CPP,BPP 的中性糖侧链虽然有较高的占比,但是HG 结构的大量降解和甲氧基、乙酰基的损失会造成Mw 的降低。前人关于甜菜果胶多糖提取的研究中也发现碱提甜菜果胶多糖的中性糖侧链含量高于酸提,而分子质量较低[37-38]。
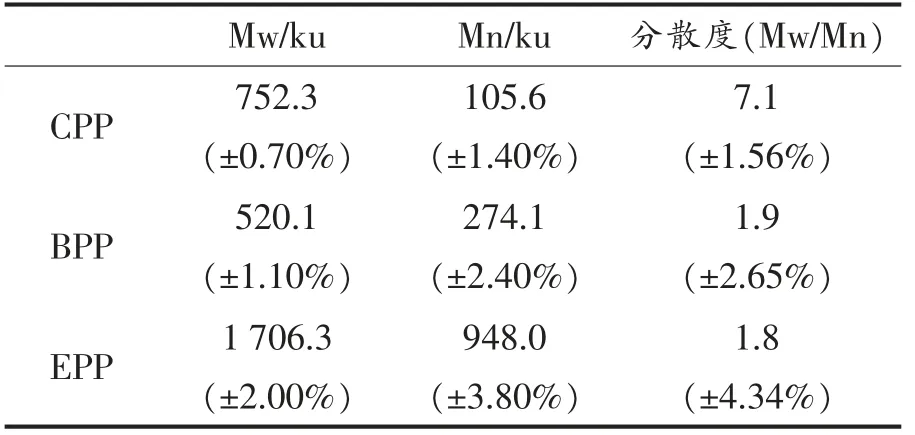
表2 不同马铃薯渣果胶多糖的平均分子质量和分散度Table 2 Average values of molecular weights and polydispersity of different potato pulp pectic polysaccharides
Mw/Mn 用来表示聚合物的分子量分散度(Polydispersity),理论上呈单分散的聚合物Mn/Mw 为1,而Mw/Mn 越高,表示分子质量分布越宽[4]。三者中,CPP 的Mw/Mn 最高为7.1;而BPP 和EPP的Mw/Mn 较小,分别为1.9 和1.8。在Yang 等[4]的研究中,不同种类的酸提取的马铃薯渣果胶多糖的Mw/Mn 介于5.2~8.0,与本试验结果相近。根据Renard 等[25]的研究,中性糖之间的糖苷键易受到酸的裂解,因此酸提马铃薯渣果胶多糖中可能存在以中性糖为主的低分子质量果胶片段,从而造成较宽的分子质量分布。
2.5 马铃薯渣果胶多糖的分子链构象
HPSEC-MALLS-RI-VISC 体系可以计算马铃薯渣果胶多糖的分子质量与其特性黏度之间的关系,从而得出Mark-Houwink-Sakurada 方程(MHS,η=KMwα)(图2)。方程中指数α 值可以用来推断多糖分子链构象,0~0.3 时,为球状链;0.5~0.8 时为无归线团状态;1.8~2.0 则代表半刚性或棒状链[39]。测得CPP,BPP 和EPP 的Mark-Houwink-Sakurada 方程α 值分别为0.4529,0.6602,0.3385,这表明在0.2 mol/L 的氯化钠流动相中,EPP 和CPP 的分子链形态介于球状链和无规线团之间,而BPP 则为无序线团态。EPP 的α 值最小,链形态最接近于球状构象,这可以归因于其较高的分支程度,也与其较大的平均分子质量有关[40]。

图2 不同马铃薯渣果胶多糖的Mark-Houwink-Sakurada 方程Fig.2 Mark-Houwink-Sakurada equations of different potato pulp pectic polysaccharides
3 结论
本研究采用酸、碱、酶法分别从马铃薯渣中提取果胶多糖,发现3 种方法提取的马铃薯渣果胶多糖均为低甲氧基的,以富含Gal 侧链RG-I 结构为主的果胶多糖。然而不同提取方法对马铃薯渣果胶多糖得率、单糖组成、分子质量以及链形态均有影响。其中,碱提果胶多糖得率最高(23.1%)且拥有最高的RG-I 占比;酸提果胶多糖甲酯化度(7.5%)和乙酰化度(13.6%)最高;酶提果胶多糖拥有最高的分子质量(1706.3 ku),可以用于进一步的降解改性。受单糖组成、分子质量和分支程度的影响,三者在分子链形态上也存出一定差异。不同提取方法得到的马铃薯渣果胶多糖的功能性还需要进一步研究,进而建立提取方式、结构、功能性三者之间的联系。
猜你喜欢
食品与生物技术学报(2022年1期)2023-01-11
盐科学与化工(2020年9期)2020-09-23
天然产物研究与开发(2019年1期)2019-03-01
中成药(2018年8期)2018-08-29
中成药(2018年3期)2018-05-07
分析化学(2017年12期)2017-12-25
天然产物研究与开发(2016年1期)2016-06-05
中华老年多器官疾病杂志(2016年7期)2016-04-28
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
