
炎症调控因子在糖尿病视网膜病变新生血管形成中的作用
2021-08-10 09:28:38陈加玉杨明明
国际眼科杂志 2021年8期
陈加玉,滕 岩,杨明明
0引言
糖尿病视网膜病变(diabetic retinopathy,DR)是劳动人口中最主要的致盲性眼病,其中以糖尿病性黄斑水肿(diabetic macular edema,DME)最为严重。DR是由于长期高血糖引起的视网膜血管系统的病理和功能改变,被认为是糖尿病最常见且最严重的微血管并发症[1]。DME是指视网膜黄斑区组织层间积液,其可发生在DR的任何阶段,是糖尿病患者中心视力丧失的主要原因。DR的发病机制复杂(图1),截至目前仍未完全阐明。长期高血糖诱导的视网膜微血管改变、炎症及视网膜神经纤维退化是引起糖尿病患者视力丧失的主要原因。此外,越来越多的证据显示,免疫机制在DR发病中起着重要作用。
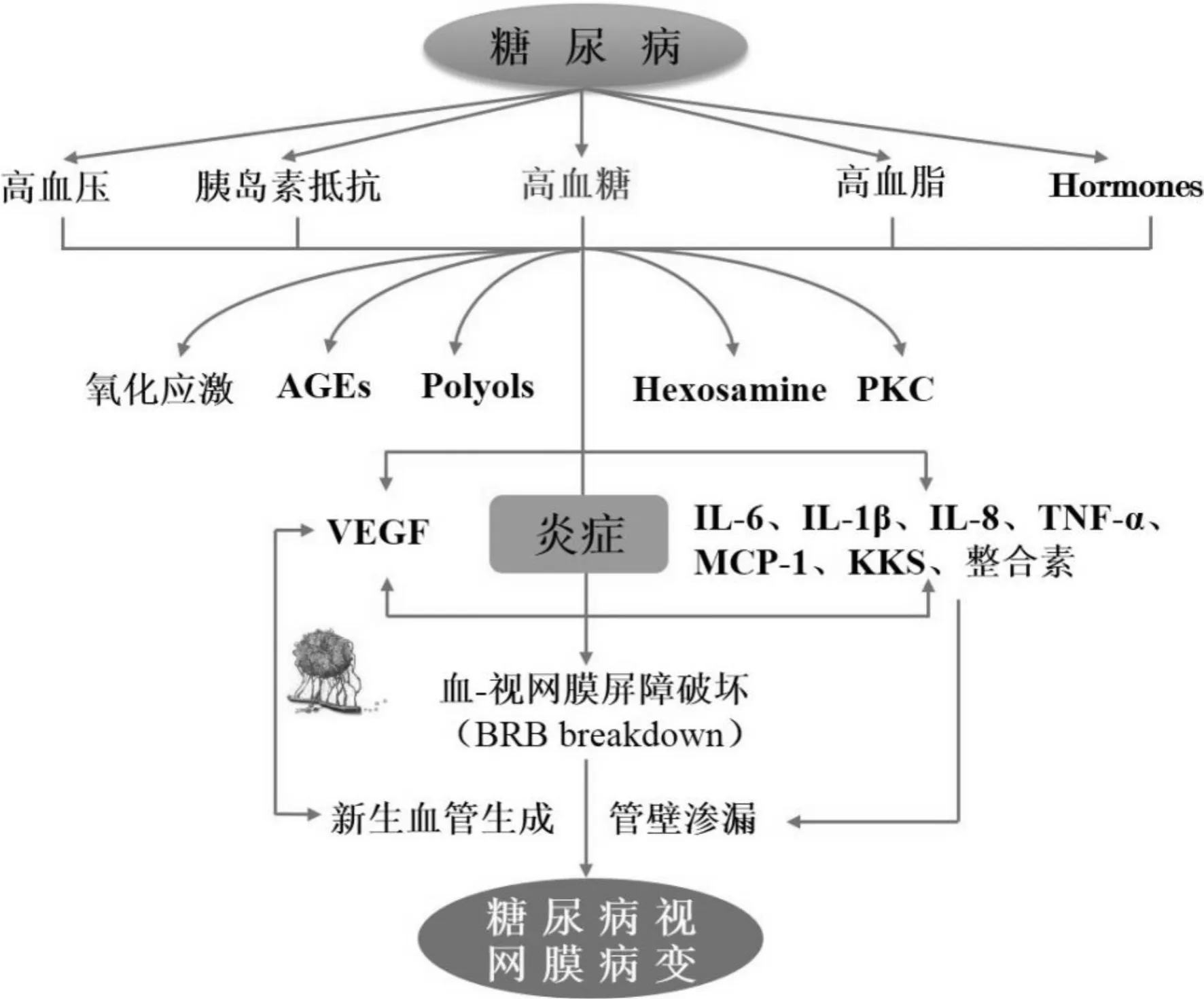
图1 糖尿病视网膜病变的发病机制。
眼球是人体具有高度组织性和复杂性的精细器官。由于视网膜内外屏障参与免疫隔离,确保了每一个腔室具有无菌的环境。生理条件下,它们能阻止细胞和大分子物质自由进出眼睛。当视网膜的生理机能发生改变时,免疫反应将迅速被激活,内环境的稳定及功能将会得到维持。然而,糖尿病患者机体在慢性炎症反应的持续刺激下,会导致严重的组织重构和功能缺失。这种低水平的慢性炎症反应称为副炎症或非经典炎症。因此,抑制这种持续的病理性炎症是治疗DR的一个新方向。最新研究表明,在DR患者眼内,参与炎症反应的细胞因子如白细胞介素(interleukin,IL)-6、IL-1β、单核细胞趋化蛋白-1(monocyte chemotactic protein 1,MCP-1)、肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)的水平显著升高[2],它们相互作用,促进局部病理性新生血管的生成,极大地促进了DR的进展。本文旨在讨论炎症在DR发病机制中的作用及其与新生血管的关系。
1新生血管相关因子
1.1血管内皮生长因子血管内皮生长因子(vascular endothelial growth factor,VEGF)由多种相关蛋白组成,包括VEGF-A、B、C、D、E和胎盘生长因子。VEGF-A是VEGF家族的典型成员,已经证明其是血管生成的关键,能够结合高亲和力酪氨酸激酶受体(VEGF-R1、R2)发挥作用,在炎症和新生血管形成中增加血管壁通透性[3]。因此,抗VEGF活性的相关药物在临床被广泛用于治疗DR和DME。缺氧是促进VEGF基因表达的最具特征的因素,但晚期糖基化终末产物(advanced glycation end products,AGEs)、IL-1β、IL-6等促炎症因子释放均可上调VEGF mRNA的表达。这些发现解释了在某些DME患者中,即使没有缺氧的证据,VEGF也会过度表达。VEGF-A通过与VEGF-R2结合,对血管内皮细胞具有促进增殖和渗出的作用。VEGF-R2激活后,酪氨酸激酶受体活性增加,导致下游多种信号通路激活,进而损害视网膜内外屏障,这也是DME发病过程中一个重要的病理机制[4]。此外,VEGF-A的浓度增加还可以上调细胞间黏附因子-1(intercellular adhesion molecule-1,ICAM-1)的表达,促进白细胞淤滞与细胞因子的释放,进而增加新生血管生成与炎症因子的表达。上述病理改变均是DR病理过程的重要标志。
1.2血管生成素血管生成素(angiopoietins,Ang)是一类生长因子,与血管内皮酪氨酸激酶受体Tie-2结合,具有调节血管稳态,控制血管通透性、抑制炎症及血管生成等功能[5],它在DR的炎症过程中具有特殊意义。Ang1与Tie-2结合,激活的信号通路通过募集周细胞起到稳定血管,同时降低由炎症因子诱导的血管通透性。相反,在高血糖、缺氧或氧化应激等不利条件下,Ang2竞争性地与Tie-2结合,抑制Ang1信号通路,导致血管稳定性降低、周细胞脱落、视网膜屏障破坏、炎症加重。Rangasamy等[6]研究表明,Ang2通过改变内皮细胞连接的VE-钙黏蛋白对视网膜内屏障产生破坏,这表明Ang2/Tie-2通路有望成为DME替代治疗的新靶点之一。
2炎症相关因子
随着研究的深入,人们发现细胞因子和其他炎症介质导致的持续低水平炎症是DR及DME发展的重要机制,其中IL-6、IL-1β、IL-8、TNF-α、ICAM-1、血管细胞黏附小分子1 (VCAM-1)、整合素b-2(CD-18)、MCP-1、血浆激肽释放酶-激肽系统(kallikrein kinin system,KKS)等已被报道在DR的发病过程中发挥关键作用[7](表1)。

表1 糖尿病视网膜病变中相关炎症因子及可能参与机制
2.1IL-6 IL-6是一种重要的细胞因子,其在宿主抵抗感染和组织损伤等环境应激方面起着关键作用。在调节失衡的情况下,IL-6的持续分泌与各种慢性炎症甚至癌症的发生有关[8]。在不同靶细胞中,IL-6具有广泛的生物学活性,包括急性期蛋白合成、调节先天和获得性免疫反应等。
IL-6信号通路参与内皮细胞功能障碍和血管炎症,在DR和其他炎症性眼病的发病中起着重要作用。IL-6是VEGF介导的炎症性血管渗漏的重要介质,可以通过直接阻断内皮细胞和上皮细胞的屏障作用,或诱导其他细胞因子的表达,促进血管通透性增加[9-10]。Valle等[10]报道IL-6通过上调内皮细胞特异性ICAM-1的表达,导致人视网膜微血管内皮细胞的屏障破坏。此外,Yun等[11]另一项体外研究表明,IL-6可以诱导转录激活因子3(activator of transcription 3,STAT3)活化,进而下调紧密连接蛋白1(zonula occludens-1,ZO-1)和闭锁蛋白,增加内皮细胞的通透性,ZO-1和闭锁蛋白是形成视网膜内屏障细胞间紧密连接的主要成分。此外,IL-6也被发现通过诱导VEGF的产生促进视网膜新生血管的形成。
临床发现,许多DR患者玻璃体和房水中IL-6浓度升高,预示炎症因素在其中发挥重要的作用。一项临床研究中显示,IL-6受体抑制剂托珠单抗(tocilizumab)对药物难治性黄斑水肿有显著疗效[12]。此外,一项前瞻性研究表明,在接受雷珠单抗治疗的DME患者中,房水中IL-6浓度基线水平较低的治疗组预后视力更好[13]。表明IL-6的浓度具有指导治疗的作用,可能成为抗VEGF治疗效果的候选生物标志物。
2.2IL-1β IL-1β是一种主要的促炎症细胞因子,通常在感染、组织损伤或免疫失衡时产生[14]。它在介导自身炎症综合征、心血管疾病和癌症进展方面发挥着重要作用。在眼内,IL-1β可以诱导视网膜内屏障的许多变化,包括白细胞募集、通透性增加、内皮细胞形态和功能的改变。研究发现,Müller细胞是视网膜中IL-1β细胞的主要来源,高血糖诱导的caspase-1/IL-1β信号通路激活,是诱导细胞焦亡的起始过程[15]。IL-1β除了具有强的促炎症作用外,还可诱导血管生成。值得注意的是,IL-1β和VEGF在内皮细胞中相互上调,对诱导促血管生成反应至关重要[16]。多项临床研究表明,DR/DME患者眼中IL-1β水平升高。IL-1β抑制剂卡纳单抗可显著改善增殖性糖尿病视网膜病变患者血管渗漏及黄斑水肿程度[17]。此外,IL-1β拮抗剂阿纳金拉不仅显著降低了激光诱导脉络膜新生血管鼠模型中视网膜下新生血管的发育[18],而且也能改善DR患者及实验动物的内皮功能[19]。
2.3IL-8 IL-8是一种促炎症趋化因子,属于CXC趋化因子家族,被认为是中性粒细胞、单核细胞和淋巴细胞的重要激活剂和趋化剂,也是新血管形成的重要介质。研究表明,难治性DME患者房水中IL-8水平更高[20]。表明其能够将中性粒细胞和单核细胞募集到玻璃体中,在视网膜新生血管形成过程中起到关键作用。IL-8浓度与DR引起的黄斑水肿的严重程度呈正相关,而与视网膜分支静脉阻塞引起的黄斑水肿无相关性[21],说明IL-8在DME的发生发展中亦起着重要的作用。Liu等[22]研究证实IL-1β可诱导IL-8表达显著增加,这可能进一步增强DR患者中IL-1β的致病作用。这说明DR炎症反应过程中不同细胞因子和趋化因子之间存在明显的交互作用。
2.4TNF-α TNF-α是一种促炎症细胞因子,与免疫介导、心血管和肿瘤疾病均有关联。TNF-α具有多种生物学效应,包括细胞增殖、分化、死亡等,其被认为与DR发病机制及眼内炎症等密切相关[23]。大量体内及体外研究表明,TNF-α增加白细胞对视网膜内皮细胞的黏附,增加视网膜内屏障的通透性[24]。Jo等[25]研究表明,在高脂饮食和链脲霉素诱导的糖尿病大鼠中,视网膜色素上皮中的小胶质细胞分泌TNF-α,激活核转录因子-κB(nuclear transcription factor-κB,NF-κB),从而降低ZO-1的分布。同时,使用糖皮质激素可以抑制NF-κB信号通路,阻止TNF-α诱导的视网膜内皮细胞通透性增高。此外,依那西普可以有效降低糖尿病大鼠TNF-α表达,抑制NF-κB的活化,减少视网膜渗漏及视网膜细胞凋亡[26]。临床药物研究表明,TNF-α抑制剂英利昔单抗(infliximab)可有效改善DME患者预后视力[27],提示TNF-α阻断治疗可作为DR治疗的潜在药物靶点之一。
2.5MCP-1 MCP-1也称CCL2,是CC趋化因子家族成员之一。MCP-1可以刺激单核细胞和巨噬细胞的募集及活化,引起视网膜血管渗透性改变、活性氧形成、细胞损伤和炎症反应,在DR的发病机制中起到重要作用[28]。此外,这种趋化因子通过促进VEGF的产生,在血管生成及纤维化中亦起到关键作用。值得注意的是,MCP-1的表达也受NF-κB的调控。临床研究显示,DR患者眼内液中MCP-1表达水平更高,表明MCP-1是在DR患者的局部视网膜上生成的[29]。此外,玻璃体腔液中MCP-1浓度与DR的严重程度具有显著相关性[28]。这说明MCP-1有可能成为预示DR严重程度的生物标志物。
2.6其他KKS在血管损伤过程中被激活,在炎症反应、血流变和凝血中具有重要作用。临床研究表明,晚期DR患者玻璃体腔液中构成KKS的成分血浆激肽释放酶(plasma kallikrein,PKal)含量增加[30]。激活眼内KKS系统可引起视网膜血管通透性改变、血管舒张和视网膜增厚,进而导致黄斑水肿。药物临床试验表明,一种新型的双环PKal抑制肽可有效改善糖尿病小鼠的视网膜通透性及炎症性损伤[31]。上述研究表明KKS成分是DME潜在的治疗靶点,目前针对该通路的临床试验正在进行中。
另一个参与糖尿病炎症级联反应的靶点是整合素,整合素家族是介导细胞-细胞和细胞-细胞外基质相互作用的细胞黏附受体。它具有多种生物学活性,如细胞分化、黏附、迁移和增殖等。众所周知,DR患者白细胞与视网膜微血管系统的黏附显著增加,通过中性粒细胞抑制因子的表达,选择性拮抗这种黏附,已被证明可以逆转糖尿病引起的视网膜微血管病变[32]。一项临床研究显示,白细胞整合素αmβ2(也称为CD11b/CD18或MAC1)是一种介导白细胞与内皮细胞黏附的蛋白,它通过激活白细胞促进视网膜内皮细胞的损坏[33]。因此,预防白细胞黏附视网膜内皮细胞可能成为治疗DR的潜在干预靶点。
3炎症与新生血管的关系
血管生成和炎症在许多病理和生理条件下关系十分密切。炎症细胞可产生血管生成因子、生长因子和蛋白酶等,它们有助于在炎症部位形成新的血管结构。同时,新生血管的内皮细胞可以促进白细胞招募和促炎分子的产生。新生血管和炎症有许多共同的信号通路和分子介质,环氧合酶/前列腺素便是其中一种。此外,许多促炎细胞因子(如IL-1α、IL-1β、IL-6、TNF-α、IL-8)可直接或间接作用于血管内皮细胞产生促血管生成介质,诱导血管形成[34]。此外,VEGF和Ang1可能通过上调细胞黏附分子和炎症因子的表达引起血管内皮细胞的炎症反应。
炎症在DR的发病机制中起着重要作用,特别是在早期阶段,通过激活转录因子和上调促炎症/促血管生成因子的表达发挥作用[35]。炎症和血管改变在DR中密切相关。促炎症细胞因子、趋化因子和其他炎症介质导致的慢性炎症可导致视网膜血管损伤、视网膜病理性新生血管和黄斑水肿。此外,炎症可能导致DR患者视网膜神经变性和小胶质细胞激活,促进血管生成因子和细胞因子的产生[36]。在DR患者中,炎症与新生血管相互促进,极大地增加了病情的严重程度,针对DR的抗炎与抗新生血管治疗依旧是DR患者药物治疗的重点。
4结论与展望
DR是一个全球性的公共健康问题,也是导致失明的主要原因。目前已明确长期慢性炎症反应是DR发生发展的重要机制。抗VEGF药物的发现是治疗DR的一个重大突破,但单一抗VEGF治疗存在局限性,一种新型的双特异性单克隆抗体药物法瑞西单抗(faricimab)应运而生,它能同时结合VEGF-A和Ang2,具有高亲和力和特异性[37]。针对DME治疗的Ⅱ期临床试验结果显示,与雷珠单抗治疗组相比,法瑞西单抗可进一步减少中心区视网膜厚度,有效改善DR疗效。多种炎症介质的发现有助于揭示DR发病机制中的炎症要素及环节,也为新型药物研发及联合治疗提供了可能。目前,针对其他抗炎靶点的药物,如整合素或细胞因子阻滞剂等,正在进行早期临床试验,这些新的抗炎化合物可能比传统类固醇药物更具选择性和安全性,这使它们更适合用于DR和DME等慢性疾病的治疗。希望有朝一日,在深入了解DR发病机制的基础上,开展精准医疗,探索靶向性炎症调控对疾病进程的影响,为DR的早期防治及临床诊疗提供新思路、新靶点。
猜你喜欢
现代仪器与医疗(2022年2期)2022-08-11 09:53:56
中国慈善家(2022年1期)2022-02-22 21:39:45
中医眼耳鼻喉杂志(2021年1期)2021-07-22 07:38:28
中医眼耳鼻喉杂志(2021年2期)2021-07-21 08:53:34
中国眼镜科技杂志(2019年9期)2019-11-11 12:15:32
海峡姐妹(2017年7期)2017-07-31 19:08:23
安徽医科大学学报(2016年12期)2017-01-15 14:21:48
湖南中医药大学学报(2016年1期)2016-12-01 04:08:18
中国病理生理杂志(2015年8期)2015-12-21 12:38:16
视野(2015年4期)2015-07-26 02:56:52
