
氟骨损害相关信号通路及其交互调控作用
2021-08-07 05:33刘纯周星宇杨世榕李仲伟贾莹
环境与职业医学 2021年7期
刘纯,周星宇,杨世榕,李仲伟,贾莹
贵州医科大学口腔医学院,贵州 贵阳 550004
氟是自然界广泛分布的元素之一,适宜量的氟对人体具有益处,它参与人体骨骼和牙齿的生长,但当人体氟含量超出正常值,则会造成氟蓄积甚至氟中毒。全球25 个国家,超过2 亿人有氟中毒风险。氟中毒可对牙齿、骨骼、软组织以及神经系统等造成损害,影响人体美观、生理功能甚至丧失正常生活能力[1-3]。氟对骨的生成与吸收具有双向性特征:既可以加强成骨活动,造成骨硬化,又可促进骨吸收,导致骨质疏松;但总体而言,主导作用是促进成骨活动增强[4]。氟暴露后,机体内各类因子发挥作用,相应的信号通路发生改变,引起成骨细胞和破骨细胞增殖分化、活性和功能的改变,从而表现出不同的骨相损害。这一疾病表现受多条信号通路的多维调控,因此对氟中毒信号调控机制开展研究,对明确相关疾病的发病机制、制定相应防治措施至关重要。本文将对近年氟化物对骨组织在不同信号通路的影响及信号间串扰形成的网络交互调控作用进行阐述。
1 氟对骨组织信号通路的影响
1.1 刺猬(hedgehog,Hh)信号通路
Hh 信号首先在果蝇中发现。高等脊椎动物体内编码Hh 蛋白的基因有三种,分别是音速刺猬因子(sonic hedgehog,SHh)、沙漠刺猬因子(desert hedgehog,DHh)、印度刺猬因子(Indian hedgehog,IHh)。Hh 信号通路既参与软骨祖细胞的定型、增殖与分化的调控,也参与成骨细胞分化,影响正常骨组织的形成[5]。Hh 信号通路的核心组成部分包括分泌型糖蛋白配体Hh、两种主要的膜蛋白受体—即跨膜蛋白受体(patched,Ptch)和跨膜蛋白(smoothened,Smo)、锌指转录因子(gliomaassociated oncogene homolog,Gli)及下游目的基因。当Ptch 与SHh 配体结合后,它对Smo 的抑制作用被解除。当Hh 缺乏[6],Hh 配体不与其受体Ptch 结合时,Ptch 对Smo 起抑制作用。这时,Smo 下游全长的Gli 转录激活子受多种激酶作用而发生磷酸化,以羧基端被截短形式进入细胞核内,被蛋白酶体剪切成截短的转录抑制子形式,抑制SHh靶基因转录。当Hh存在时[7],Hh 与Ptch 相结合,Ptch 对Smo 抑制作用则解除。全长Gli 蛋白进入核内,激活下游转录基因,促使软骨细胞、成骨细胞增殖分化。由此可见,整个信号通路中,Hh 配体及Smo 所起到的是正调节作用,而Ptch 则发挥负调节作用,转录因子Gli 能够反映Hh 信号通路活化的程度[8]。此外,近年发现,Hh还存在非经典通路,包括:(1)由Ptch 启动但不依赖Smo 蛋白信号通路,通过调节细胞周期蛋白(cell cycle protein,Cyclin)在细胞内的定位发挥对细胞周期的调控作用;(2)依赖Smo 蛋白直接激活信号通路,可通过激活ras 同源家族成员A(ras homolog family member A,RhoA)调节多种细胞的肌动蛋白细胞骨架,进而调节细胞的运动和迁移;(3)不依赖Hh 配体、Ptch 以及Smo,由Gli 蛋白直接激活信号通路三种模式[9]。
研究表明,氟中毒大鼠模型多器官氟损伤的发生与Hh 信号通路表达改变相关。在氟中毒大鼠肝脏组织中,可以检测到SHh和Gli1基因的mRNA 表达高于对照组,并且在大量肝脏实验中均可发现以上相关蛋白的表达[10]。不仅肝脏,在软骨组织中也有同样表达,且随染氟浓度改变而变化。邓超男等[11]对大鼠进行染氟处理,同时加以腹腔注射Hh 信号通路特异性阻断剂环巴胺,发现随染氟浓度增加,大鼠软骨组织中SHh、Smo、Gli1 表达逐渐增强,而注射阻断剂的大鼠骨组织Gli1 表达则较单纯染氟大鼠明显降低,大鼠出现不同程度的软骨骨化及细胞凋亡等关节软骨损伤,其机制可能与Hh 信号通路蛋白表达改变及软骨细胞凋亡有关。同样的结果在成骨细胞培养过程中以氟化物和环巴胺进行干预的实验中得以验证[12]。以上实验提示Hh信号通路可调控软骨的增殖、分化,干预软骨和骨的形成,而氟离子可通过调节SHh 和Gli等因子异常表达来调控Hh 信号通路,导致氟中毒的发生。
Rho激酶(rho-associated coiled-coil-forming kinases,ROCK)是小G 蛋白Rho 相关激酶,被认为是Rho/ROCK信号通路的最核心成员,使用氟化钠能够显著增强大鼠股骨中ROCK-1 的基因和蛋白表达,表明Rho/ROCK参与了氟化钠诱导的骨细胞凋亡[13]。在氟暴露的人群中,随着氟含量的增加,Cyclin D1 的mRNA 转录和蛋白表达逐渐增加。经氟化钠处理的人成骨细胞中,Cyclin D1 的表达同样增加,促进了氟诱导的成骨细胞活化[14]。因此,有理由推测,氟同样可以通过Hh非经典信号通路调控成骨细胞活化及凋亡,促进氟骨症的发生。
1.2 Wnt/β-连环蛋白(β-catenin)
Wnt 信号通路是一条保守信号传导通路,由经典及非经典两类通路组成,有19 个成员已被确认。经典Wnt 信号通路为Wnt/β-catenin 途径,调控干细胞的多能分化、器官的发育和再生,对胚胎骨骼的发育及出生后骨的生长具有至关重要的作用[15]。Wnt 是该信号通路的关键启动因子,核心因子β-catenin 主要位于细胞膜,而在胞浆中游离量较少。缺乏Wnt 时,β-catenin 在支架蛋白、糖原合成酶激酶3β(glycogen synthasc kinase-3β,GSK-3β)等形成的复合体的作用下被磷酸化,最后被泛素化降解。当Wnt 存在时,低密度脂蛋白受体相关蛋白5/6(low density lipoprotein receptor related protein 5/6,LRP5/6)、Wnt 和膜受体卷曲蛋白形成的复合体将GSK 从磷酸化β-catenin 转移至LRP5/6,使得β-catenin 聚集,从而胞浆内得以稳定存在,然后进入胞核后,与T 细胞因子/淋巴增强因子(T cell factor/lymphoid enhancer factor,TCF/LEF)相互作用促进靶基因表达[16-17]。但在该通路的初始阶段,分泌性糖蛋白1(dickkopf-related protein 1,Dkk1)能够在细胞外就与Wnt蛋白辅助受体LRP5/6结合,从源头上阻断Wnt/β-catenin 通路。
有研究表明长时间暴露于高氟环境可导致Wnt通路的细胞外拮抗剂Dkk1 降低,Wnt/β-catenin 信号被激活,GSK-3β 活性降低,β-catenin 的表达增多,加速了骨基质分泌成骨细胞的进程,证明了Wnt/β-catenin 信号可能调节成骨细胞的增殖和成熟[18-19]。Zeng 等[20]对196 名高氟地区暴露者进行采样分析,发现随着氟暴露量的增加,Wnt 信号抑制剂Dkk1 的含量逐渐降低,而GSK-3β、β-catenin 的浓度和成骨分化的重要生化指标Ⅰ型胶原A1 与骨碱性磷酸酶的浓度却增加,同样表明氟可以通过减少Wnt 信号抑制因子Dkk1 的表达来激活Wnt 信号,从而促进骨形成;但GSK-3β 在以上两次实验中出现截然相反的结果,猜测可能是信号通路中、下游的因子存在负反馈调节而导致的。Chu 等[21]、Zhan 等[22]的研究提供了体内和体外证据,证明氟化物可诱导Wnt/β-catenin信号的异常激活,促进β-catenin 在细胞核内的聚集,积累的β-catenin 进入细胞核并与LEF1 形成复合物,从而激活一系列靶基因的转录,增加小鼠松质骨形成和Wnt3a、GSK-3β 和Runt 相关转录因子2(runtrelated transcription factor 2,Runx2)蛋白表达;当抑制β-catenin 时,氟诱导成骨同样表现出抑制现象,揭示了氟诱导成骨细胞异常激活的潜在机制,验证了β-catenin 是介导成骨细胞活力和分化的中枢分子,其在细胞质中的积累和核定位对Wnt/β-catenin 信号通路的激活至关重要。
1.3 Notch
Notch 通路是一条进化高度保守的信号通路,一种在间充质/成骨细胞谱系中调节自我更新和分化的细胞间通信通路。哺乳动物中有4个同源受体和5个同源配体,其中同源受体是Notch1~4,都是Ⅰ型跨膜蛋白,均由胞内区(Notch intracellular domain,NICD)、跨膜区和胞外区(Notch extracellular domain,NECD)组成。Notch同源配体(Delta/Serrate/LAG-2,DSL)都是Ⅰ型跨膜蛋白,分为Delta like与Jagged-1/2。Jagged-1是一种有效的骨诱导蛋白,可积极调节动物创伤后骨愈合。Notch 信号转导无须第二信使和蛋白激酶的参与,可直接接收邻近细胞的信号并传到细胞核,激活相关转录因子的表达。当NICD 未释放时,核结合蛋白(CBF1 suppressor of hairless-lag1,CSL)是一个转录抑制因子;当Notch 受体接收到相邻细胞膜上的DSL 信号后,Notch 受到两种连续的蛋白裂解影响,去整合素金属蛋白酶10(a disintegrin and metalloprotease 10,ADAM10)或肿瘤坏死因子-α转换酶(TNF-α converting enzyme,TACE)释放出NECD,然后γ 分泌酶释放出NICD,NICD 进入胞核与CSL 蛋白形成复合物,将原来的“协同抑制复合物”转换为“协同活化复合物”,形成活化复合物并驱动下游靶的转录[23]。
Notch 受体是细胞命运和功能的决定因素,调节成骨细胞和破骨细胞分化,并在骨骼发育和骨骼重塑中起着核心作用[24-25]。陈修文等[26]在大鼠饲氟6 个月后,取骨进行检测,发现高氟组大鼠的Notch、Jagged-1 蛋白水平表达明显呈下降表现,而且与染氟剂量呈相关性,推测过量氟会通过抑制Notch、Jagged-1 来增加成骨细胞的分化,从而表现出骨质硬化。氟与Notch 通路关系的研究还处于空白区域较多的阶段,有待学者进一步深入了解其在基因和蛋白质水平的作用。
1.4 核因子κB(nuclear factor-κB,NF-κB)受体激活子(receptor activator of the nuclear factor-κB,RANK)/ NF-κB受体活化因子配体(NF-κB receptor activator ligand,RANKL)/骨保护素(osteoprotegerin recombinant protein,OPG)
RANK 是RANKL 的唯一受体,是肿瘤坏死因子受体家族中的成员,是一种I 型跨膜蛋白,RANK 通过与RANKL 结合发挥调节骨吸收的关键作用。OPG 作为一种分泌型糖蛋白,由成骨细胞分化,能够以二聚体的形式竞争性结合RANKL,使RANKL 丧失与RANK 结合的功能性,抑制破骨细胞分化成熟,使骨代谢向着骨硬化方向发展,RANKL/OPG 浓度的比值调节破骨细胞的形成[27-28]。巨噬细胞集落刺激因子(macrophage colony-stimulating factor,M-CSF)和RANKL 对于启动破骨细胞分化必不可少,在M-CSF 的介导下,RANKL结合在破骨细胞前体的受体RANK,从而启动细胞内的信号转导,促进破骨细胞成熟与分化[29]。
RANK/RANKL/OPG 骨转换轴被认为是骨改建过程中调解骨吸收的“终极通路”。孙秀娟等[30]建立大鼠氟中毒模型组中,M-CSF 明显高于其他组,证明氟化物能够促进骨吸收,但具体的机制没有进行说明。也有学者研究报道,用环肽F可以干扰M-CSF和RANKL信号的共同影响因子原癌基因c-Fos,M-CSF诱导分化阶段难以分化破骨细胞,但RANKL相较而言诱导较易[31]。将RANKL 的诱饵受体OPG基因敲除,小鼠的颞骨切片在光镜下观察,出现了局灶性高细胞区域,显示出骨吸收和沉积,并且含有多核破骨细胞[32]。从分子层面来看,建立骨细胞-破骨细胞共同培养模型后,发现随着氟浓度的增加,转化生长因子β1 表达升高,OPG 竞争性抑制得到加强,从而抑制破骨细胞分化和成熟,骨吸收被减弱,导致氟骨症的发生[33]。由此可见RANKL 才是诱导破骨细胞分化,促进骨吸收的关键因子,M-CSF 只是起阶段性介导作用。在氟干预下M-CSF、RANKL对骨吸收的调控得到增强,而随着氟浓度的增加,OPG的竞争性抑制逐步表现出优势。
1.5 甲状旁腺激素(parathyroid hormone,PTH)
PTH 是甲状旁腺分泌的维持体内钙稳态的激素,其分泌主要受血浆Ca2+浓度的调节。PTH 主要与PTH受体(parathyroid hormone-related protein,PTHrP)结合而发挥其生物功能,PTH I 型受体是II 型G 蛋白偶联受体,在骨组织中,能够表达PTH I型受体的细胞主要为成骨细胞系[34]。多项研究以证实,氟化物增加骨对PTH 作用的抵抗力,PTH 及其PTH I 型受体在氟骨症中具有重要影响,成骨细胞和成骨细胞前体表面有PTH I 型受体,因此PTH 可以直接作用;而破骨细胞表面没有相应受体,只能够通过成骨细胞和骨细胞来进行间接作用[35]。氟中毒常继发甲状旁腺功能亢进,当机体缺钙时,PTH 分泌增多,氟化物增加骨骼对PTH 作用的抵抗力,可能降低降钙素分泌,从而引发代偿性PTH 活性[36-37]。PTH 的效应取决于细胞暴露在PTH 的周期性和剂量大小:间歇性低剂量给予PTH,可以促进成骨细胞分化和矿化,骨量得以增加;持续暴露于高剂量PTH 时,骨骼产生分解,骨吸收占主导[38]。PTH 对骨影响的作用机制已经在临床上得到应用,在骨质疏松的治疗中取得良好疗效。
2 交互调控
信号的激活或抑制不是仅在其本身信号通路内的分子中以线性方式完成的,而是多条信号通路成员或分子相串扰,形成庞大而复杂的效应网络,以协调或拮抗的方式控制细胞内的各种生物过程,交互调控(cross-talking),共同完成细胞生理功能[39]。
Hh 信号主要在软骨膜和关节软骨中表达,由于Wnt 信号也在软骨成骨中具有重要意义,为了验证两者关系,研究人员检测双基因突变小鼠的Gli1等靶基因,证明了Hh 信号在Wnt/β-catenin 信号上游,并且经过成骨细胞相关基因表达的检测,证明了Wnt 在Hh 下游[5,40]。猜测可能是由于Hh 通路中Wnt 抑制因子-1(Wnt inhibitory factor 1,Wif-1)的出现,抑制了Wnt/β-catenin 通路,使β-catenin 降解无法聚集,靶基因无法完成转录[41]。从骨形成角度来看,膜内骨化过程,Hh 信号作用并不明显;软骨内骨化过程则Hh 信号水平较高,可控制软骨细胞肥大和成骨细胞分化,当Hh 和Wnt/β-catenin 维持在一定水平,骨可以正常形成[42]。这也能够体现出两条信号通路存在串扰,但骨形成涉及的通路甚是复杂,此次实验的结果究竟是以何种形式所造成,并无具体结果加以说明。实验数据表明Wnt 家族成员Wnt 能够通过促进OPG 的产生来抑制破骨细胞的生成,而Wnt 信号抑制因子Dkk 的过表达降低了Wnt、β-catenin 蛋白的水平,从而促进了RANKL/OPG 值和成骨细胞的凋亡;Dkk 通过影响成骨细胞增殖,成骨细胞前分化为成熟成骨细胞和新的骨基质形成来调节骨形成过程[43-44]。
Sun 等[45]建立了PTH基因敲除小鼠和野生型小鼠股骨骨折模型,利用实时聚合酶链反应和蛋白质免疫印迹等技术检测内源性PTH 的功能和机制,发现PTH 敲除组的RANKLmRNA 表达均低于野生组,说明其对破骨细胞的影响是间接产生的,PTH 是通过磷脂酰肌醇3 激酶/活化丝氨酸-苏氨酸蛋白激酶B/转录活化子(phosphoinositide-3-kinase/serine-threonine proteinkinase B/signal transducer and activator of transcription 5,PI3K/AKT/STAT5)信号通路来促进成骨细胞分泌RANKL,从而影响破骨细胞的数量和功能,这一系列试验共同说明PTH 是促进骨吸收的。Yu 等[46]的研究支持这一结论。PTH 被证明是硬骨素(sclerosteosis,SOST)的抑制剂,在经典Wnt 通路的上游,SOST 可以抢占LRP5/6 复合体的结合位点,直接阻断这一通路[47]。由此可说明Wnt 经典通路在PTH和Hh 信号通路中都在下游起作用[48]。将氟和PTH 作为共同处理因素,与单独氟处理相比,PTH 和氟共同处理的分组SOST 和Dkk1 得到了反向增加表达,同时在这项研究中,RANKL 蛋白的表达水平上调,降低了OPG 表达,这导致RANKL/OPG 值显著增加[26,49]。染氟之后,PTH 相关蛋白等相关分子的表达显著增加,PTHrP 对IHh表达的抑制作用增强,从而使IHh表达下降,抑制了IHh/PTHrP反馈环,抑制了软骨内骨化[50]。
有研究显示,RANKL在破骨细胞分化过程中诱导了Jagged-1和Notch2表达。而Notch2 NICD的异位表达促使活化T-细胞核因子1活性增强,OPG/RANKL比例降低,诱导的破骨细胞生成。这些结果表明Notch 信号在OPG/RANKL 的下游,活化T 细胞核因子是RANKL刺激Notch2-NF-κB 复合体的直接作用靶点,Notch 通过逐级信号协调NF-κB 间接激活破骨细胞[51]。但是该实验中有许多矛盾之处,需要进一步的研究来确定Notch 信号传导的分子机制在破骨细胞分化中的作用。Notch 抑制Wnt 活性,即使β-catenin 水平的正常化也不足以完全逆转此作用,这是因为Notch 胞内域NICD 诱导了多毛/增强子-1(hairy/enhancer of split-1,HES-1)的表达增强。而HES-1 RNA 干扰实验表明,HES-1 在NICD 对Wnt/β-catenin 信号传导的抑制作用中起着核心作用,HES-1 的参与抑制了Wnt/β-catenin 信号传导和成骨细胞生成[52]。在Rallis 等[53]的研究中,观察到Jagged-1 表达和Gli1 表达之间具有统计学意义的相关性,由于Gli1 是Hh 通路激活的主要效应分子,这种关联可能表明两条通路之间可能存在串话,这一概念需要进一步研究。Notch 信号可能在上游功能控制Hh 反应,在斑马鱼脊髓模式形成过程的实验中,抑制Hh 信号不影响Notch 通路活性,但Notch 信号的激活导致Hh 通路活性增强;对Notch 信号的抑制消除了脊髓中Hh 依赖和Hh 独立的Gli1 表达,Notch信号允许神经祖细胞通过Gli转录调控和Gli蛋白维持对Hh 信号做出反应[54]。但这一表现主要存在于脊髓模式,在成骨细胞中是否也是同样机制还未可知。图1为氟对Hh、Wnt/β-catenin、Notch、RANKL/OPG和PTH 通路的影响及通路之间交互调控网络的示意图。
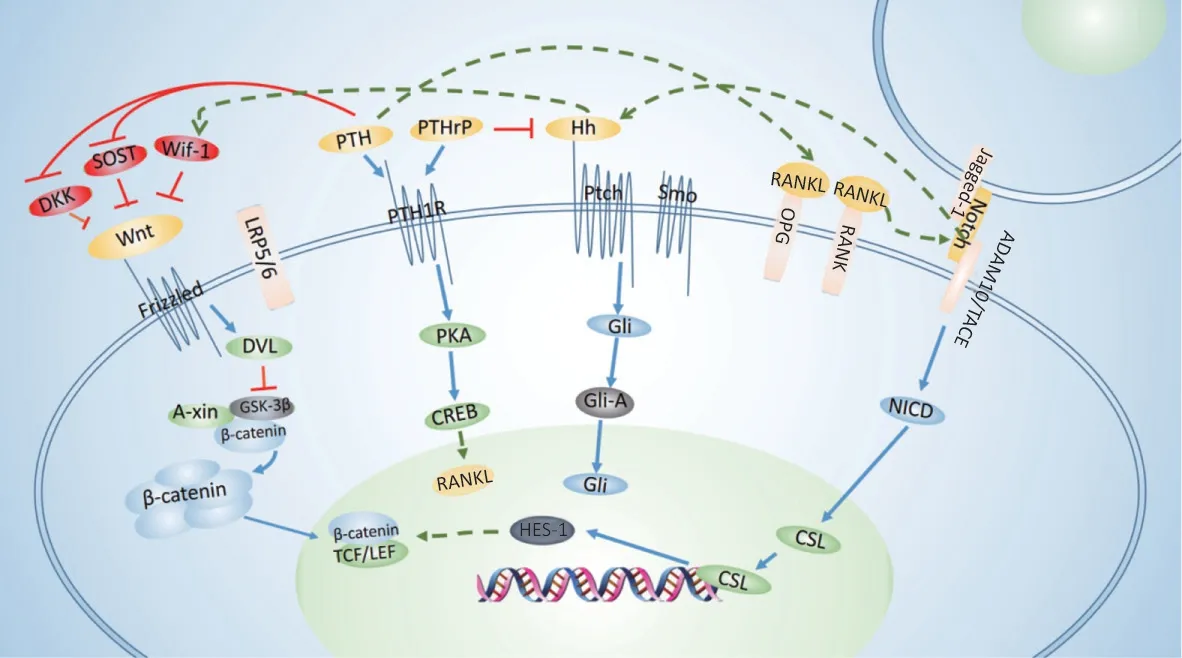
图1 氟对Hh、Wnt/β-catenin、Notch、RANKL/OPG和PTH通路的影响及通路之间交互调控网络的示意图Figure 1 A schematic picture of the effects of fluoride on Hh,Wnt/β-catenin,Notch,RANKL/OPG,and PTH signaling pathways and the cross-talks among the pathways
3 展望
Hh、Wnt/β-catenin、Notch、RANKL/OPG 以及PTH代表着调节生长发育的基本途径。迄今为止,对于这些信号通路在慢性氟中毒诱导骨转换的分子机制主要都集中在信号通路的线性研究上。然而,许多研究表明,各信号通路的发生并非孤立的开展,而是相互串扰,交互调控。骨的形成或吸收,依靠信号通路间的协同效应、竞争性抑制或者反馈调节,通过专注氟骨损害相关信号通路网络连接点的分子,深入剖析它们之间的联系,了解氟中毒的发病机制,有可能开发出更高效更具有特异性的靶向治疗策略。
猜你喜欢
中国骨质疏松杂志(2021年9期)2021-10-08
中国临床医学影像杂志(2019年5期)2019-08-27
中国临床医学影像杂志(2019年1期)2019-04-25
中国临床医学(2019年3期)2019-01-04
安徽医科大学学报(2016年12期)2017-01-15
中国民族医药杂志(2016年6期)2016-05-09
罕少疾病杂志(2016年4期)2016-03-11
中国骨质疏松杂志(2016年1期)2016-01-29
中国医科大学学报(2015年10期)2015-03-01
中国医学科学院学报(2012年3期)2012-03-25
