
Breast cancer: Muscarinic receptors as new targets for tumor therapy
2021-08-02 05:42:46AlejandroEspaolAgustinaSalemYamilaSanchezMarElenaSales
Alejandro Español, Agustina Salem, Yamila Sanchez, María Elena Sales
Alejandro Español, Agustina Salem, Yamila Sanchez, María Elena Sales, Laboratory of Immunopharmacology and Tumor Biology, CEFYBO CONICET University of Buenos Aires,Buenos Aires C1121ABG, Argentina
Abstract The development of breast cancer is a complex process that involves the participation of different factors. Several authors have demonstrated the overexpression of muscarinic acetylcholine receptors (mAChRs) in different tumor tissues and their role in the modulation of tumor biology, positioning them as therapeutic targets in cancer. The conventional treatment for breast cancer involves surgery, radiotherapy, and/or chemotherapy. The latter presents disadvantages such as limited specificity, the appearance of resistance to treatment and other side effects. To prevent these side effects, several schedules of drug administration, like metronomic therapy, have been developed. Metronomic therapy is a type of chemotherapy in which one or more drugs are administered at low concentrations repetitively. Recently, two chemotherapeutic agents usually used to treat breast cancer have been considered able to activate mAChRs. The combination of low concentrations of these chemotherapeutic agents with muscarinic agonists could be a useful option to be applied in breast cancer treatment, since this combination not only reduces tumor cell survival without affecting normal cells, but also decreases pathological neo-angiogenesis, the expression of drug extrusion proteins and the cancer stem cell fraction. In this review, we focus on the previous evidences that have positioned mAChRs as relevant therapeutic targets in breast cancer and analyze the effects of administering muscarinic agonists in combination with conventional chemotherapeutic agents in a metronomic schedule.
Key Words: Muscarinic receptors; Drug therapy; Breast cancer; Drug combination;Metronomic therapy; Drug resistance
INTRODUCTION
Cancer is a heterogeneous disease characterized by the loss of normal behavior of cells and the acquisition of new characteristics that lead to malignant transformation. These characteristics include high growth and division rate, and the ability to invade neighboring tissues and to disseminate to distant organs to generate metastases[1].
For many years, researchers thought that tumors had a clonal origin[2,3], but taking into account the high intra-tumor heterogeneity observed, they later considered the coexistence of different cell subpopulations within a tumor. One of these subpopulations,known as cancer stem cells, was identified in 1994 in an acute myoleid lymphoma[4]and later described in several solid tumors such as lung, breast, colon, prostate and brain tumors[5-8]. These cells have self-renewal and differentiation capacity, and also exhibit high expression of drug extrusion pumps, the latter of which confers them resistance to chemotherapy[9]. In addition, several authors have described that this cell population is responsible for the failure of response to cancer treatment and for cancer recurrence[10-12].
Also, it has been demonstrated that primary and metastatic tumor cells present phenotypic, genotypic and epi-genotypic differences[13]. These differences result in several changes in the expression and function of membrane protein receptors as well as in their signaling pathways. Thus, this intra-tumor heterogeneity creates a great challenge in the selection of specific biomarkers and treatments in oncology[14].
The transformation of a normal cell into a tumor one is a complex and progressive process in which the cell acquires genetic modifications like deletions or other mutations in tumor suppressor genes and/or oncogenic genes. Since these genes control cell proliferation either directly or indirectly, deletions or mutations in these genes may allow tumor cells to grow without control, disseminate and invade other tissues[15].
Tumor suppressor genes can either inhibit the cell cycle or promote apoptosis. The loss of their functionality can cause the development of cancer, as demonstrated in ovarian[16], lung[17], colorectal[18], head and neck[19], pancreatic[16], uterine[20],osteosarcoma[21], gastrointestinal[22], bladder[23] and breast[24] tumors.
The presence of several of these genes is associated with a high incidence of cancer.These genes can be divided into highly and moderately penetrant in relation to cancer.Highly penetrant genes includeBRCA1,BRCA2,TP53,PTEN,CDH1,STK11andPALB2, whereas moderately penetrant ones includeCHEK2, ATM,BARD1,BRIP1,NBN,NF1,RAD51DandMSH6[25-27].
Oncogenes, on the other hand, are abnormal or mutant genes related to normal genes called proto-oncogenes, which support tumor development. From the beginning of the 1980’s, the participation of these genes has been studied and associated with tumor progression[28]. Many studies have demonstrated that proto-oncogenes affect the activity of telomerase enzyme in different tumor cell types. Due to the latter, these cells exhibit short telomeres and chromosomal instability[29]. Somatic mutations in the telomerase gene promoter have been described in gliomas[30], head and neck cancer[31], thyroid carcinoma[32], hepatocellular carcinoma[33], squamous cell carcinoma[34], bladder cancer[35], breast carcinoma[36] and melanomas[37].
In an incipient stage of tumor growth, when malignant cells exert a rapid rate of proliferation, the nutritional requirements of cells increase, leading to an angiogenic switch. The latter implies the development of new blood vessels from the ones preexisting in the tumor environment, which allows the emerging tumor to duplicate its diameter from 1 mm to 2 mm[38]. Concomitantly, changes in the cellular biological properties lead tumor cells to invade the surrounding extracellular matrix, to intravasate into the nearby blood and lymphatic vessels, and then to disseminate through the circulation to distant organs and to metastasize[39].
According to data from the International Agency for Research on Cancer, in 2020 this illness produced more than 19 million new cases and caused almost 10 million deaths worldwide. In particular, breast cancer has the highest incidence among all female cancer types. It represents 24.5% of the total new recorded female cancer cases and is one of the main causes of death in women, corresponding to 15.5% of deaths from this disease (Figure 1)[40].
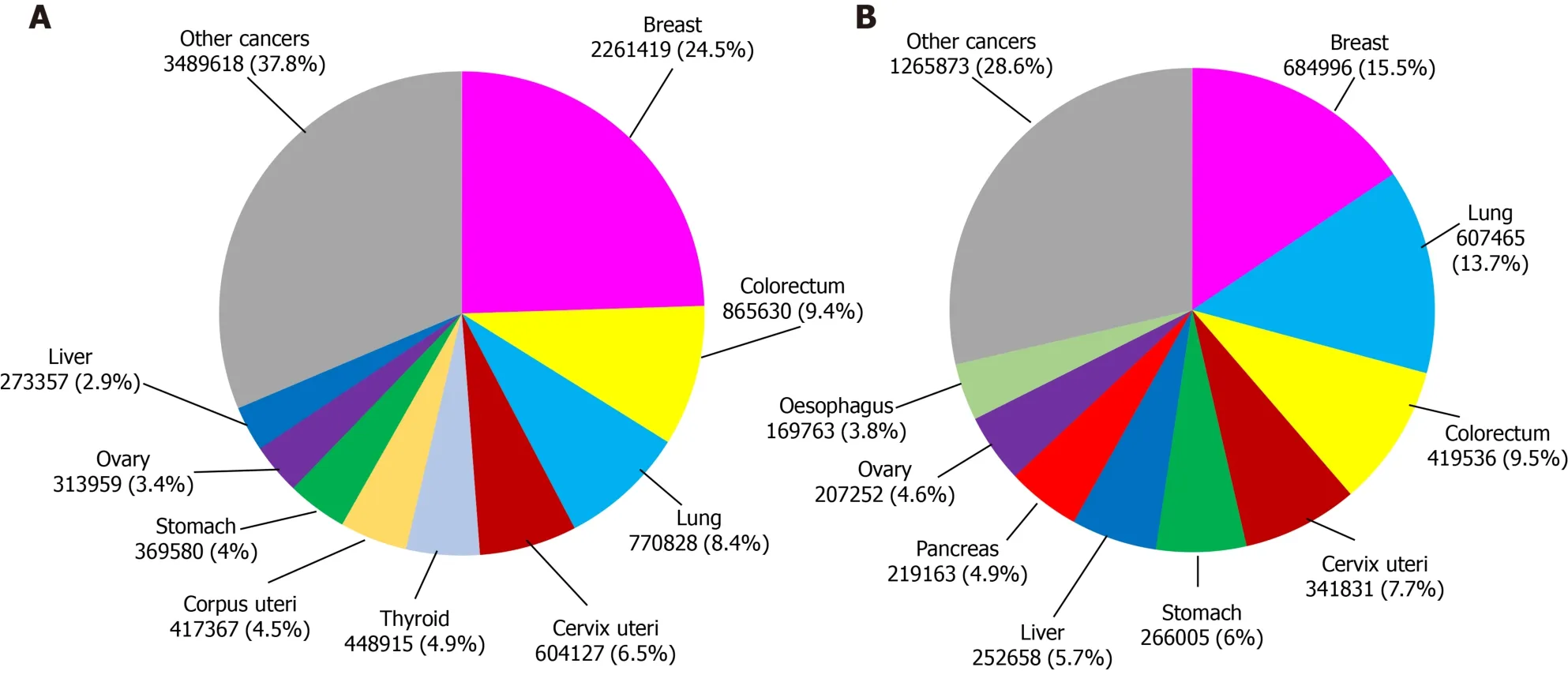
Figure 1 Female world incidence of new cases of different types of cancer and number of cancer deaths in 2020. A: Different types of cancer;B: Number of cancer deaths.
Since life expectancy in the world population has become higher, the risk of developing tumors has also increased, considering that this parameter increases with age[41]. It is expected that the number of new cases of breast cancer in 2040 will rise to 3.2 million people and there will be more than 1 million deaths unless treatments present higher effectiveness[40].
BREAST CANCER
The breast is composed of lobules (which are milk-producing glands), the ducts that connect the lobules to the nipple, and connective, fatty and lymphatic tissues. Breast cancer occurs when there is an uncontrolled growth of cells within any of these components. Although tumors can appear in any tissue of the breast, it occurs most frequently in the lobules[42].
Breast tumors exhibit particular histopathological and biological characteristics that require specific and different antitumor strategies. For these reasons, the adequate classification of tumors has a main therapeutic importance[43-45].
The different types of therapies against breast cancer include surgery, radiation and the administration of immunobiological or chemotherapeutic agents[46,47].
Surgery (tumorectomy or mastectomy) is one of the main options of treatment for patients at different stages of this disease, whereas radiotherapy is used as a complement to surgery and/or chemotherapy to reduce the probability of tumor relapse. The combination of tumorectomy and radiotherapy is used as a replacement for mastectomy in patients at the first stages of the disease.
Chemotherapy consists of the use of drugs to kill tumor cells. It can be administered as adjuvant therapy, when drugs are applied after another treatment like surgery in order to eradicate tumor cells that might have survived. Oncologists also administer neoadjuvant chemotherapy before surgery to reduce the tumor size and to be more likely to completely eliminate the tumor after the procedure[40].
As described below, according to their mechanism of action, chemotherapeutic agents can be classified into: endocrine drugs, immunological agents, DNA alkylating agents, antimetabolites and antimitotic drugs[48].
Endocrine drugs are used to treat estrogen receptor (ER) or progesterone receptor(PR) positive breast tumors. These drugs are synthetic analogs of the anti-gonadotropin-releasing hormone, anti-progestins or anti-estrogens. The latter can be divided into aromatase inhibitors and ER antagonists. The most common side effects of these drugs are: flushing sensation, nightly sweats, vaginal dryness, high blood clot risk,apoplexy, cataracts, endometrial cancer, uterus cancer, bone-mass decrease and gastrointestinal symptoms[40,49].
Immunological agents are drugs that can stimulate the patient´s immune system to detect and eliminate breast cancer cells. Immunological agents also include monoclonal antibodies against specific tumor cell proteins, and cancer treatment vaccines. Different types of immunological agents can be administered to inhibit check-points, block the suppression of the immune response and/or over-activate Tcells from patients. The most common side effects of these drugs are: fatigue, fever,shivers, weakness, nausea, vomits, dizziness, body aches and high or low blood pressure[40,50-52].
Regarding DNA alkylating agents, the ones used to treat breast cancer include cyclophosphamide and doxorubicin. Cyclophosphamide, which is an oxazaphosphorine, interferes with the duplication of DNA and RNA transcription, and its most common undesired effects are: myelosuppression, hepatotoxicity, pulmonary fibrosis,nephrotoxicity, mucosis, megaloblastic anemia, birth defects and neurotoxicity[40,53].
Doxorubicin, which is an anthracycline, is a topoisomerase II inhibitor that increases DNA degradation, preventing cell replication. Additionally, it can intercalate into DNA and promote the formation of free radicals, which in turn causes the fragmentation of DNA strands. Unfortunately, this agent can also cause cardiotoxicity,myelosuppression, alopecia, extravasation, infertility and urine discoloration[40,53].
The antimetabolites used in breast cancer can be divided into antifolates (such as methotrexate) and pyrimidine antagonists (such as 5-FU, capecitabine or gemcitabine).Methotrexate is a competitive inhibitor of the dihydrofolate reductase which causes a decrease in pyrimidine and DNA synthesis. On the other hand, 5-FU or capecitabine can form a complex with the thymidylate synthase-folic acid, inhibiting its activity and reducing thymidine and DNA synthesis, whereas gemcitabine can incorporate a pyrimidine analog into DNA, decreasing its synthesis. The most common collateral effects of these drugs are: myelosuppression, cardiotoxicity, hepatotoxicity, pulmonary toxicity, hemolytic uremic syndrome and hyperammonemic encephalopathy[40,53].
Finally, the antimitotic drugs used in breast cancer treatment can be divided into three types: vinca alkaloids (such as vinorelbine), taxanes (such as paclitaxel and docetaxel) and non-taxane microtubule inhibitors (such as eribulin, ixabepilone and epothilone). Vinorelbine binds to B tubulin, inhibiting its polymerization into microtubules; it also prevents the formation of the mitotic spindle and arrests cells in the M phase. Taxanes produce hyper-stabilization in polymerized microtubules, thus inhibiting the degradation of the mitotic spindle, and also arrest cells in the M phase.Non-taxane microtubule inhibitors can either prevent the formation of the mitotic spindle (eribulin) or bind to B tubulin, hyper-stabilizing microtubules and producing cell arrest. Undesirable actions of these drugs are: myelosuppression, peripheral neuropathy, QT prolongation and hypersensitivity[40,54-56].
Selecting the most effective chemotherapeutic agent to treat breast tumors requires the determination of their genetic profile. Tumors are classified according to the immunohistochemical analysis of the protein expression levels of ER, PR, human epidermal growth factor receptor type 2 (HER2) and Ki-67. This analysis allows defining four breast cancer subtypes: (1) Luminal A; (2) Luminal B; (3) HER2 positive;and (4) Basal-like or triple negative[57,58].
Luminal A tumors express ER and/or PR, are HER2 negative and present low levels of Ki-67 protein, which modulates tumor cell growth[59]. This tumor subtype is the most frequently diagnosed annually and has a 5-year relative survival rate of 94%[60].The conventional treatment for these tumors is endocrine therapy[61].
Luminal B tumors express ER and/or PR and can be positive or negative for HER2 expression. This tumor subtype has a higher cell proliferation rate than the luminal A subtype because it expresses a higher concentration of Ki-67 protein[61]. Luminal B tumors represent 10% of the total of annually diagnosed breast tumors and have a 5-year relative survival rate of 90%[60]. This tumor subtype is usually treated with endocrine therapy and in some cases with HER2 targeting drugs or other chemotherapeutic agents[61,62].
HER2 positive tumors have a high expression of this molecule and do not express ER or PR. These tumors also have a high concentration of the Ki-67 protein[59]. This tumor subtype represents 5% of all annually diagnosed breast tumors and has a 5-year relative survival rate of approximately 80%[60]. HER2 positive tumors are usually treated with HER2 targeting antibodies like trastuzumab[61] or margetuximab[63,64]alone or combined with other chemotherapeutic agents[65,66].
Triple negative tumors do not express ER, PR or HER2, but express a high concentration of Ki-67 protein and are usually very invasive and agressive[59]. This tumor subtype represents 10% of all annually diagnosed breast tumors and has a 5-year relative survival of approximately 80%[60]. These tumors can be subdivided into basal,claudin-low, normal-like and other less frequent subtypes[43,67], and, since they do not have a specific therapeutic target, they do not have any specific treatment.Although some authors indicate the use of a platinum-based agent such as cisplatin,the results are not encouraging due to the low life expectancy of the patients[68,69].
The 5-year relative survival rate varies significantly from patients with luminal A tumors, who have the best prognosis, to patients with triple negative tumors, who have the worst prognosis. The decrease in the survival rate observed in triple negative tumor patients can be explained by different factors, including higher resistance to chemotherapy and/or radiotherapy, higher relapse probability, and higher ability to metastasize in comparison with other tumor subtypes[70].
Although in recent years the diagnosis, classification and treatment of breast cancer have improved, treatment failure, recurrence and mortality are still reported worldwide.
It must be taken into account that, one of five women worldwide will develop cancer during her lifetime and one of eleven will die from this illness[40].
MUSCARINIC ACETYLCHOLINE RECEPTORS
Muscarinic acetylcholine receptors (mAChRs) are G protein-coupled receptors(GPCRs), which belong to the superfamily of seven-transmembrane domain receptors and can modulate many functions in normal and tumor cell biology[71,72]. Regarding the latter, several authors have described an increase in the expression of different GPCRs in tumor tissues. GPCRs include thrombin receptor[73], protease-activated receptor-1[74], angiotensin II receptor type I[75], GPR161[76], GPR81[77] and leucinerich repeat-containing G-protein-coupled receptor 5[78].
mAChRs were firstly described in the central nervous system[79] and then in the parasympathetic nervous system[80]. More recently, mAChRs, together with nicotinic receptors, have also been localized in non-neuronal cells. Also, their ligand,acetylcholine (ACh) and the enzymes that synthesize and degrade it have also been detected out of the nervous system, defining a new organization known as the nonneuronal cholinergic system[81]. In our laboratory, we described for the first time the over-expression of mAChRs of the GPCR family in breast tumor tissues and in cancer cell lines from murine and human origin[82,83].
mAChRs are metabotropic receptors and five subtypes (M1 to M5) have been identified. When activated, they can trigger different signaling pathways known as canonical or non-canonical in distinct tissues[84].
These receptors are glycoproteins with seven hydrophobic transmembrane domains connected by three extracellular and three intracellular hydrophilic loops. The domains assemble forming a structure with a pocket where the agonist binds. Also, the cytoplasmic region of the receptor couples to G protein, which is composed of three subunits: α, β and γ[85]. When a ligand binds to the receptor, guanosine diphosphate is released and replaced by guanosine triphosphate, while the subunits are dissociated into a βγ dimer and the guanosine triphosphate-bound α monomer. Depending on the α subunit type (Gαs, Gαi, Gαq, and Gα11) different downstream effectors are stimulated[86].
Agonists like ACh or the synthetic non-hydrolyzable analog carbachol can activate M1, M3 and M5 receptors, which couple to a Gαq protein, which in turn up-regulates phospholipase C activity. This enzyme cleaves phosphatidylinositol 4,5-bisphosphate into 1,2-diacylglycerol and inositol 1,4,5-triphosphate. 1,2-diacylglycerol activates protein kinase C, which stimulates downstream proteins, causing calcium influx.Inositol 1,4,5-triphosphate leads the sarcoplasmic reticulum to release stored calcium,which modulates the activation of many calcium-dependent enzymes like nitric oxide synthase[87]. Additionally, the M3 receptor can activate Ras-Raf-1-Erk-Akt through a non-canonical pathway[88].
In turn, the activation of M2 and M4 receptors, which are coupled to a Gαi/o protein, inhibits adenylyl cyclase, decreasing cyclic adenosine 3’,5’-monophosphate(cAMP) production from ATP. The decrease in cAMP concentration subsequently blocks the activation of protein kinase A[87]. M2 and M4 receptors can also regulate the activity of potassium and calcium channels[89].
The expression of the different mAChR subtypes changes throughout different body organs and tissues. This differential expression leads to diverse responses to the same stimuli. Therefore, the identification of the mAChR subtypes in different cell types is important, especially because they could have therapeutic potential. The nervous system, for example, expresses all subtypes[90-92], whereas the M1 receptor is mainly expressed in salivary glands[93], pancreas[94], bladder[95] and respiratory pathways[96]. The M2 receptor subtype is predominantly expressed in cardiac[97], digestive[98]and respiratory[99] tissues, whereas the presence of M3 receptor protein has been documented in salivary glands[93], pancreas[100], bladder[101], lung[102], colon[103]and gastric smooth muscle[104]. Finally, the M4 and M5 receptors are also expressed in the lung[105,106], while the M4 subtype is also detected in gastric tissue[107].
The expression of mAChRs in different tissues can be regulated by various stimuli.Grodzkiet al[108] demonstrated that the expression of M2 receptor can be increased by the treatment of sympathetic neurons with gamma interferon. In line with these results, at our lab, we demonstrated that thede novoexpression of M3 and M5 receptors can be induced by the treatment of NIH3T3 fibroblasts with interferon gamma plus lipopolysaccharide fromEscherichia coli, and that this leads to an increase in the sensitivity of these cells to carbachol[109].
In several diseases, including cancer, mAChRs or their signaling pathways are differentially expressed in comparison to healthy tissues. This could be useful at the moment of considering these receptors as therapeutic targets, as described in many respiratory diseases[110]. It has been demonstrated that in pulmonary arterial hypertension, for example, the administration of M3 receptor agonists induces an antihypertensive therapeutic response[111]. Additionally, in chronic obstructive pulmonary disease, it has been described that the usage of muscarinic antagonists combined with beta2 adrenergic receptors reduces hyperinflation, improves dyspnea,and reduces exacerbations while improving cardiac functions[112]. This combination of muscarinic antagonists and beta2 adrenergic receptors has also been studied as a therapeutic treatment for asthma, showing promising results[113].
Alterations in the expression of mAChRs or their signaling pathways have also been detected in several diseases of the central nervous system[114-117]. However,researchers could not determine whether these alterations are either the causes or the consequences of these pathologies[118]. In spite of this, muscarinic therapy has shown promising results for these diseases. It has been documented that the treatment of patients with Alzheimer or Huntington diseases with M1 receptor agonists causes beneficial effects, reducing the symptoms of these diseases[119,120]. Furthermore, the usage of M1 antagonists in Parkinson's disease and multiple sclerosis leads to positive results, also reducing the disease symptoms[121,122].
Different authors have determined that the expression of mAChRs in malignant tissues is different from that in normal tissues. These differences comprise an increase in the expression of receptors and/or a modulation in the subtype expression pattern.In human colon cancer, for example, the expression of the M3 receptor subtype is increased more than 100-fold respect to normal tissue and its activation modulates cell proliferation, progression and invasion of this neoplasia[123]. Also, in small cell lung cancer, the M3 receptor subtype is up-regulated, promoting cell migration and invasion[124]. In human bladder, while normal tissue expresses M1, M2 and M3 receptors, tumor tissue expresses only the M2 receptor and its activation induces a decrease in cell proliferation and migration[125]. Regarding breast cancer, several studies from our laboratory have demonstrated the presence of different mAChR subtypes in breast cancer cells of murine and human origin that promote tumor growth and angiogenesis, and the absence of these receptors in normal mammary cells[126-129].
The predominant expression of different mAChR subtypes in human tumors is summarized in Table 1.

Table 1 Summary of the expression of muscarinic acetylcholine receptor subtypes (M) in different human cancers
It has been reported that the activation of mAChRs leads to the stimulation of several steps of tumor progression, involving different receptor subtypes in each tumor. Cell proliferation has been found to be induced by the M5 receptor subtype in melanoma cells[130], by the M1 and M4 receptor subtypes in glioblastoma cells[131],by the M3 receptor subtype in stomach and colon cancer[132,133], by the M2 receptor subtype in non-small cell lung cancer[134] and by all receptor subtypes in theesophagus[135].
In other types of tumor, researchers have observed not only an increase in cell proliferation, but also an increase in cell invasiveness and migration ability. This increase is induced by the M1, M3, M4 receptor subtypes in prostate tumors, by the M1, M3, M4 and M5 receptor subtypes in cervical tumors[136-139] and by the M1 and M3 receptor subtypes in hepatocellular carcinoma[124,140]. Finally, in oral cavity tumors, cell motility and dissemination have been found to be increased by the activation of the M4 receptor subtype[141].
Another important aspect to analyze is the expression and function of mAChRs in primary tumors and their metastases. Regarding the latter, two melanoma cell lines and a third one derived from a metastasis have been found to express M1 and M3 receptors. Although all of them responded to carbamylcholine, increasing cytosolic calcium levels, the metastatic cell line responded with a higher peak in calcium concentration. The authors speculated that this difference could be responsible for a higher malignance and migratory potential in metastasis[142]. In accordance with the latter observations, we have reported that the expression of mAChRs is significantly higher in the metastatic murine mammary cell line LMM3 than in the non-metastatic LM3 tumor cells that originated them[82]. However, when analyzing the function of mAChRs in tumor tissues, other authors confirmed opposite results, particularly considering the expression and function of the M2 receptor subtype. Lucianòet al[143], for example, established that two cell lines derived from a neuroblastoma with bad prognosis, SK-N-BE and SK-N-BE(2C), express the M2 receptor and that its activation inhibits the cell cycle. In human bladder tumors, Paciniet al[125] reported the expression of M2 receptor protein and found that its stimulation reduced cell proliferation and migration. These results are in line with the findings of Alessandriniet al[144], who demonstrated that the activation of the M2 receptor increases apoptosis in glioblastoma, a brain tumor with bad prognosis. Regarding leukemia, Cabadaket al[145] identified the presence of the M2 and M3 receptors in K562 cells, and linked their activation to an inhibition in cell proliferation. In head and neck cancer, Sunet al[146]reported a similar effect by M3 activation. Finally, in human pancreas tumor, Renzet al[147] demonstrated that M1 receptor activation decreases tumorigenesis.
Regarding breast cancer, we have reported the expression of all mAChR subtypes in the breast adenocarcinoma cell lines LM2 and LM3 derived from spontaneously aroused tumors in female BALB/c mice and the lack of expression of these receptors in the murine mammary non-tumorigenic cell line NMuMG[82]. Using tritiated quinuclidinyl benzilate ([3H]-QNB) in binding assays, we determined the expression mainly of the M2 and M3 receptor subtypes[126]. We also found that the addition of carbachol to tumor cells induces two opposite actions depending on the concentration of the agonist and the time of treatment. At low concentrations added for 1 h or less, we reported a stimulation in cell proliferation, migration and also in tumor angiogenesis,whereas at higher concentrations or longer periods of treatment, we observed cell death[126]. These differences due to distinct experimental conditions besides the expression of different mAChRs could explain the opposite results documented in the literature previously mentioned.
Luminal breast cancer tumors are characterized by the expression of their endocrine receptor. In MCF-7 cells, the human luminal breast cancer cell line most studied in oncology research, we identified the expression of the M3 and M4 receptors by Western blot. We also found that the short-time treatment of MCF-7 cells with carbachol activates, through the M3 receptor, a phospholipase C/protein kinase C/calcium-dependent nitric oxide synthase signaling pathway, increasing cell proliferation. Carbachol also stimulates the formation of tumor blood vessels and the expression of vascular endothelial growth factor A, concomitantly with tumor cell migration and matrix metalloproteinase-9 expression and activity[148,149]. Similarly to that reported in murine normal mammary cells, the non-tumorigenic human breast cell line MCF-10A does not express these receptors[148].
Triple negative breast tumors are characterized by the absence of a specific therapeutic target and bad prognosis. By studying two different human cell lines derived from this type of tumor, at our lab, we documented that both express mAChRs, but while MDA-MB231 cells express the M1, M2, M4 and M5 receptor subtypes, MDA-MB468 express all receptor subtypes including M3 protein[128].Similarly to that documentated in our previous reports, short-time activation of these cell lines with carbachol stimulated proliferation in a dose-dependent manner. This effect was prevented by the presence of the muscarinic antagonist atropine, confirming the involvement of mAChRs in this effect[128]. In MDA-MB231 cells, we were able to identify the receptor involved in the mentioned effects by using small interfering RNA silencing assays, which revealed that the one responsible for the proliferative actions in this cell line is the M2 subtype[128].
Based on previous reports by other authors and our results proving that long-term and low-concentration treatment with muscarinic agonists induces a decrease in cell viability, we next decided to focus on mAChRs as therapeutic targets for breast cancer treatment in triple negative tumors[128,129].
To assign more specificity to mAChRs as blanks of action in anti-tumor therapy, it must be taken in mind that non-tumorigenic human mammary cells, like MCF-10A, or breast samples from patients with benign pathology (fibroadenoma) lack expression of mAChRs and, as a consequence, they are not sensitive to the treatment with muscarinic agonists. However, when transfected with mAChRs, normal cells acquire the ability to respond to muscarinic treatment[129]. All these results highlight the relationship between mAChRs and breast cancer development and treatment.
CHEMOTHERAPEUTIC DRUGS INTERACTING WITH MACHRS
Many chemotherapeutic drugs are used in breast cancer treatment, but only two of them are able to bind with the active site of mAChRs: paclitaxel[82] and doxorubicin[150]. Both induce an inhibitory effect on cell proliferationin vitrosimilar to that observed with the cholinergic agonist carbachol[128].
Paclitaxel
Paclitaxel is a drug of first choice in the treatment of breast cancer. It is a taxane derived from the treeTaxus brevifolia[151]. This drug was approved by the Food and Drugs Administration (FDA) for the treatment of ovarian cancer in 1992, for advanced stages of breast cancer in 1995[152], and for the early stages of breast cancer in 2001[153]. Paclitaxel is a diterpenoid pseudoalkaloid made up of an N-benzoyl phenylisoserine group and a taxane ring. In contrast with other taxanes, paclitaxel has a lateral complex chain connected to its taxane ring in C-13, which gives this drug its antitumor activity[154]. Its molecular formula is C47H5NO14and its chemical structure is described in Figure 2.

Figure 2 Chemical structure of paclitaxel.
Despite the extended use of paclitaxel in anti-tumor treatment, it has been reported that its administration at therapeutic concentrations (10-6mol/L approximately) causes adverse effects like hepatotoxicity, leukopenia, neutropenia, anemia, thrombocytopenia, neurotoxicity, vomits, alopecia, fatigue, mucositis and diarrhea[155-158]. In addition to the low-efficiency synthesis of paclitaxel and its side effects, another problem is its insolubility in water, which affects its bioavailability. Thus, to improve its availability, it is dissolved in polyoxyethylated castor oil as a vehicle (Cremophor EL); however, this oil has been associated with several anaphylactic reactions like dyspnea with bronchospasm, hypotension and urticaria since it activates the complement system[159]. Because of this, new technologies have been developed to improve the biodistribution of this taxane, and alternative ways of administration like paclitaxel binded to albumin[160,161], paclitaxel-loaded PEG-vitamin E nanoparticles[162,163] and micellar paclitaxel[164,165] have been proposed.
The conventional dose of paclitaxel administered to treat breast cancer is 175 mg/m2skin by intravenous inoculation for 3 h every 21 d in 6 cycles[166] and its halflife time after inoculation is 4.9 ± 3.6 h[167]. After one administration, most of the paclitaxel in the circulation binds to proteins and its systemic clearance is 700 mL/min on average[168]. Approximately 90% of the drug is degraded by hepatic metabolism by P-450 isoenzymes (CYP3A and CYP2C) and is excreted by feces, while the other 10% is excreted by urine without alterations[168-170].
Paclitaxel exerts its anti-tumor effects through different mechanisms. One of these is by acting as a cytostatic drug that binds to the β subunit of tubulin and stabilizes the polymerized microtubules, inhibiting their depolymerization[171,172]. Consequently,cells lose their ability to divide due to insufficient requirements in the G2/M mitotic control checkpoint[173]. The prolonged arrest of mitosis eventually leads to cell death[174].
Besides its antimitotic effect, paclitaxel also acts in the immune system since its administration is associated with the change in the macrophage phenotype from an M2 to an M1 profile through TLR4 (toll-like receptor 4) activation[175]. Millrudet al[176] reported that, in the presence of paclitaxel, primary human monocytes turn into proinflammatory M1 macrophages, contributing to tumor eradication.
Also, at low doses, paclitaxel can induce apoptosis in tumor cells by inactivating the B-cell lymphoma 2 protein (Bcl-2)[177-179]. Additionally, it has been described that this cytostatic drug modulates several non-coding RNAs that have a regulatory function over genes related to tumor progression[180]. Moreover, paclitaxel can modulate the release of the apoptogenic factor cytochrome that can trigger programmed cell death[181,182]. The dysregulation of these and other parameters can lead to resistance to paclitaxel treatment.
One interesting point that we have analyzed in our laboratory is the ability of paclitaxel to interact with mAChRs in a specific manner[126]. We proved that paclitaxel displaces the binding of [3H]-QNB to mAChRs expressed in murine mammary adenocarcinoma cell lines in a manner similar to that of atropine. Moreover,when we treated these cells with paclitaxel at very low concentrations (10-11mol/L),we reported an inhibitory effect on tumor cell proliferation. This effect was prevented by the previous treatment of cells with atropine, confirming the participation of mAChRs in this action[126].
Doxorubicin
The other chemotherapeutic drug frequently used in breast cancer treatment is doxorubicin[183,184], which is an antibiotic that belongs to the family of anthracyclines and is produced by the bacteriumStreptomyces peucetius. It was approved by the FDA in 1974 for metastatic breast cancer treatment[185]. Its molecular formula is C27H29NO11and its chemical structure is shown in Figure 3.

Figure 3 Chemical structure of doxorubicin.
The treatment with doxorubicin causes many side effects, including cardiotoxicity[186], infertility[187], genotoxicity[188], amenorrhea[189], thrombophlebitis[190] and lung embolism[191]. Regarding cardiotoxicity, the additional challenge is that it can occur 10 years after chemotherapy treatment, appearing as a progressive congestive cardiac failure secondary to a non-ischemic dilated cardiomyopathy, and is irreversible and usually fatal[192,193]. Although many authors indicate that this effect is mediated by cardiac adrenergic interactions[194-196], Chugunet al[197] pointed out that it would be due to muscarinic interactions by modulating the ionotropic effect of carbachol.
In breast cancer, doxorubicin is administered in 60/75 mg/m2per dose intravenously in one inoculation every 21 d. Its half life time is approximately 48 h[198] and its systemic clearance is between 700 and 1250 mL/min[199,200]. Most of the doxorubicin is degraded by hepatic metabolism and the unaltered drug and its metabolite are excreted mainly by the gallbladder and in a small fraction by the kidneys[201].
Doxorubicin exerts its chemotherapeutic effect by three mechanisms. The first one is the inhibition of the enzyme topoisomerase II-α, which regulates the superhelical state of DNA and has a structural function regulating the tension of the DNA strands[202].Doxorubicin stabilizes the binding of topoisomerase II-α to the cleaved DNA,preventing the replication process and leading the cell to apoptosis[203]. Given that tumor cells present a higher proliferative rate, the expression of topoisomerase II-α is high, producing a greater selectivity of doxorubicin for these cells[204].
The second mechanism of action also affects directly the DNA strands. Doxorubicin intercalates into DNA, inhibiting the activity of topoisomerase II-α, which in turn causes the inhibition of DNA synthesis[205]. Moreover, doxorubicin intercalation causes the break of the DNA in double-strand fragments and chromatin condensation,which leads to an increase in apoptosis[206]. This antibiotic can also intercalate in RNA, inhibiting the activity of RNA polymerase, although it has more affinity for DNA[207].
Finally, the third mechanism of action of doxorubicin is the production of free radicals. Doxorubicin can act as an electron acceptor, transforming its quinone to a semiquinone-free radical, which causes oxidative damage and induces the cleavage and degradation of DNA. This is a mitochondrial reaction catalyzed by the enzyme cytochrome P450 reductase in the presence of NADH dehydrogenase[208]. It has also been described that the free molecular iron can interact with doxorubicin, forming toxic free radicals and reactive nitrogen species that increase nitrosative stress and mitochondrial dysfunction, promoting apoptosis[209].
Interestingly, doxorubicin can also interact with mAChRs. It has been demonstrated that doxorubicin displaces the [3H]-QNB binding in the left atrial muscle of guinea pig hearts in a concentration-dependent manner, similarly to atropine, indicating that doxorubicin can also bind to mAChRs[150,197]. Additionally, in triple negative human tumor cells, we determined that doxorubicin exerts an inhibitory effect on proliferation, comparable to that induced by carbachol[128].
Multidrug resistance
One of the most important causes in the failure of chemotherapy not only in breast cancer treatment but also in other cancers is the appearance of resistance. It can appear as a primary form, when it is present before the treatment, or acquired, when it develops after the exposure to a drug[210,211].
To improve the efficacy of chemotherapeutic agents in the treatment of breast cancer, it is important to define the mechanisms underlying resistance in this type of tumor. In breast cancer, resistance is multifactorial and implies different mechanisms and genes that exert their effects either together or separately, leading to a reduction or an inhibition of the effect of different drugs.
Regarding the factors that modulate the resistance to paclitaxel or doxorubicin,some of them are common to both drugs and others are specific to each of them. The main factors in common are the expression of drug extrusion pumps, noncoding RNAs, Bcl-2 and p53. Drug extrusion pumps are also known as ATP binding cassette(ABC) transporters. They are located in the cytoplasmic membrane and modulate the time that the drug is present inside the cell by controlling its active transport. The expression and activity of these pumps can be modified by different factors, including the chemotherapy treatment. Amawiet al[212] reported that paclitaxel treatment exerts an increase in pump activity and expression in different breast cancer cell lines,whereas Cox and Weinman[213] reported similar results in hepatocellular carcinomas treated with doxorubicin. In both cases, the increase in the expression and activity of these pumps reduced the effectiveness of the treatment. Regarding the other factors that modulate the action of paclitaxel or doxorubicin, noncoding RNAs, also called miRNA, can modulate the expression of genes that regulate apoptosis and cell survival, increasing the sensitivity or the resistance to the chemotherapeutic treatment[214-216], whereas Bcl-2 and p53 expression proteins regulate tumor growth and are important tumor markers that can be modulated by chemotherapeutic drugs,increasing the resistance to treatment[217,218].
Among paclitaxel resistance factors, the most relevant are changes in the cytoskeletal dynamics and kinetic degradation of the taxane structure. Several tumor cells express high levels of βIII-tubulin that are related to resistance to paclitaxel treatment[219,220]. When this expression is down-regulated, cells recover sensitivity to paclitaxel[221]. In this regard, Wanget al[222] described an increase in the expression of βIII-tubulin, which induces resistance because higher concentrations of the drug are needed to stabilize microtubules and block cell division.
Regarding the proteins that modulate cytoskeletal dynamics, many authors have established that an increase in the resistance to paclitaxel treatment is related to an over-expression of stathmin[223], septin[224], tubulin binding cofactor C[225] and BRCA1[226] proteins.
It is also known that enzymes of the cytochrome P450 subfamily 3A and 2C play a major role in the metabolism of taxane anticancer agents. The expression of these enzymes in solid tumors may thus play a role in thein situmetabolism of drugs as well, potentially affecting the intrinsic susceptibility of these tumors to taxane. An abnormal up-regulation of enzyme activity or expression reduces the halflife time of paclitaxel and as a consequence its efficacy in cancer patients[227].
Regarding doxorubicin, the expression of topoisomerase II-α and FOXO3 is mentioned as a main aspect in the mechanism of resistance to this drug. Doxorubicin exerts part of its chemotherapeutic effect by the binding to topoisomerase II-α, which produces DNA structure stabilization and induces apoptosis. Wanget al[222] reported that a down-regulation in topoisomerase II-α expression induces resistance to doxorubicin treatment in human breast tumor cells MCF-7. In human malignant breast samples, O'Malleyet al[228] demonstrated that topoisomerase II-α expression is a good marker to determine the resistance to doxorubicin since cells with higher levels of this enzyme are more sensitive to this anthracycline.
FOXO3 is a factor associated with longevity due to its antioxidant effect[229]. In this regard, Gomeset al[230] reported that the administration of doxorubicin in breast cancer patients can increase FOXO3 expression, inducing resistance to treatment and making it a marker of bad prognosis.
Other side effects
Besides the previous points, it must be taken into account that the treatment with these drugs can stimulate the development of metastases. Regarding this matter,Karagianniset al[231] demonstrated that thein vivoadministration of doxorubicin or paclitaxel to murine breast tumor bearers promotes the formation of micrometastases in the lung, which is the first step necessary to produce tumor cell intravasation and the subsequent formation of metastases. Moreover, Daenenet al[232] established that paclitaxel treatment also increases vascular endothelial growth factor receptor-1 expression in lung endothelial cells, which stimulates the adhesion of circulating tumor cells and the subsequent formation of metastases.
Additionally, conventional therapeutic treatment with doxorubicin or paclitaxel can affect normal cells, inducing cell death. We and other authors have demonstrated that,at therapeutic concentrations, paclitaxel besides reducing cell viability in human breast tumor cells MCF-7 and MDA-MB231 also causes cell death in normal mammary cells MCF-10A[128,233-235].
METRONOMIC THERAPY
The conventional treatment usually applied to cancer patients considers the administration of chemotherapeutic drugs at the maximum tolerated dose. Because of all the undesirable effects caused, patients need a long interval of time between cycles of treatment so that normal tissues can recover. The usage of the maximum tolerated dose is useful in tumors that rarely have a complex network of activating mutations,like acute lymphoblastic leukemia or testicular cancer. However, in other tumors like breast tumors, this conventional treatment is less effective mainly because these tumors permanently modulate the tumor microenvironment. In addition, as mentioned above, besides the toxicity induced by the administration of the maximum tolerated dose in breast cancer, development of resistance to treatment is frequently described[236,237].
One of the strategies to reduce side effects and the resistance to anti-tumor drugs is the administration of minimal doses of one or more chemotherapeutic agents, which,administered in a continuous regime or with short time intervals, improves the results of treatment. This strategy is known as metronomic therapy and its main aim is to reduce the toxicity and to increase the quality of the patients’ lives.
Metronomic therapy may be effective to inhibit tumor progression through different mechanisms that modulate not only tumor cell death but also the cancer stem cell population involved in tumor generation and metastasis[129,236]. Folkinset al[238]demonstrated that metronomic therapy with cyclophosphamide could reduce the growth of human glioma spheroids, a cancer stem cell marker. Viveset al[239]reported that the same treatment reduces the number of cancer stem precursor cells in human pancreatic tumor. Similarly, in our laboratory, we demonstrated that the treatment of human breast cancer cells MCF-7 with paclitaxel plus carbachol in a metronomic schedule causes a decrease in the cancer stem cell population[129].
The target of this kind of therapy is not only tumor cells but other cells of the tumor microenvironment[236,240-242]. Several authors have demonstrated that some chemotherapeutic drugs administered at low doses can inhibit the synthesis of proangiogenic factors produced by endothelial cells, which are necessary for tumor growth[236].
In addition, metronomic therapy is effective to modulate the activity of the immune system, by decreasing the number of regulatory T cells and increasing the population of cytotoxic T lymphocytes and natural killer cells[243,244].
Metronomic therapy is usually linked to repurposing drugs. The latter refers to the assignation of new uses for drugs usually administered to treat diseases other than cancer[245]. In oncology, there is an increasing interest in the prescription of noncancer drugs for cancer treatments due to the knowledge of their pharmacokinetics/dynamics and side effects, and because most of them are available at low cost[246].
Considering previous results obtained in our laboratory, we have recently proposed the administration of low doses of paclitaxel or doxorubicin combined with low doses of carbachol, a non-selective muscarinic agonist, or arecaidine, an M2 selective agonist,in a metronomic schedule to effectively reduce breast tumor cell viability[128]. As shown in Table 2, the effect of metronomic combinations is similar to that obtained with paclitaxel or doxorubicin administered at therapeutic concentrations (10-6mol/L).These results position muscarinic agonists in the spectrum of repurposing drugs.
Our results focused on the presence of mAChRs in tumor cells and their absence in normal cells, giving specificity to this type of anti-tumor therapy, and indicating that it also prevents cytotoxic actions in normal cells, which could be an indicator of reduced adverse effects (Table 2).

Table 2 Effect of metronomic chemotherapy targeting muscarinic acetylcholine receptors on breast tumors
It is important to mention that the treatment with paclitaxel plus carbachol not only reduces tumor cell viability but also prevents other important steps of tumor progression. This therapy diminishes tumor cell migration, cancer stem cellpercentage, neoangiogenesis and the expression of the drug extrusion pump ABCG2[128].
CONCLUSION
The research about new antitumor therapies with drugs that increase beneficial actions and reduce adverse effects is a challenge to improve breast cancer patients’ lives. The usage of repurposing drugs, like the muscarinic agonist carbachol, which synergizes the action of traditional anti-tumor drugs might be an alternative schedule focused on mAChRs as new therapeutic targets. The presence of these receptors at high concentrations not only in breast tumors but also in other types of tumor could help to find a more specific and less aggressive manner to treat cancer patients. On the other hand,metronomic therapy is effective to kill tumor cells without affecting normal cells and also decreases pathological neo-angiogenesis and the expression of drug extrusion proteins. The latter could prevent the appearance of resistance reported in conventional chemotherapy. Morein vivoexperiments are needed to confirm the effectiveness of this treatment in breast cancer models and to gain information to discard systemic adverse reactions.
 World Journal of Clinical Oncology2021年6期
World Journal of Clinical Oncology2021年6期
- World Journal of Clinical Oncology的其它文章
- Long-term complete response in metastatic poorly-differentiated neuroendocrine rectal carcinoma with a multimodal approach: A case report
- Phytochemically rich dietary components and the risk of colorectal cancer: A systematic review and meta-analysis of observational studies
- Impact of community-based exercise program participation on aerobic capacity in women with and without breast cancer
- Chemotherapy-induced neurotoxicity in the treatment of gynecological cancers: State of art and an innovative approach for prevention
- Imaging diagnosis of bronchogenic carcinoma (the forgotten disease) during times of COVID-19 pandemic: Current and future perspectives
- Review of 10 years of research on breast cancer patients: Focus on indoleamine 2,3-dioxygenase