
基于深度学习的多肽预测方法研究
2021-08-02 03:49:10刘全中
计算机技术与发展 2021年7期
梁 潇,吴 昊,刘全中*
(1.西北农林科技大学 信息工程学院,陕西 杨凌 712100;2.陕西省农业信息感知与智能服务重点实验室,陕西 杨凌 712100)
0 引 言
生物体内广泛分布着上万种多肽,近年来,随着科学研究的发展和对生命活动规律的深入探索,越来越多的功能性多肽分子被不断发现,部分多肽具有抗癌、抗菌、抗炎、抗病毒、穿透细胞等特性,这些特性为疾病治疗提供了重要依据[1]。
抗癌肽(anticancer peptides,ACPs)能破坏肿瘤细胞膜结构,对癌细胞增殖和迁移具有抑制作用,而对正常的体细胞基本无损伤,因此抗癌肽检测有助于抗肿瘤药物的研究[1];抗菌肽(anti-bacterial peptides,ABPs)对部分细菌、真菌、病毒有杀伤作用,其潜在的价值也受到医学界的广泛关注[2];细胞穿透肽(cell penetrating peptides,CPPs)被广泛用作药物进入细胞的运输载体[3];结合肽(surface-binding peptides,SBPs)有助于在噬菌体展示实验中建立高效的ELISA(enzyme linked immunosrbent assay)系统[4]。
具有治疗特性的多肽目前已经越来越广泛地应用于临床诊断和治疗中,因此识别这些多肽对于发现新的、高效的疾病治疗方法具有重要的现实意义[2]。传统的生物实验方法识别多肽耗时、耗力且成本高,随着高通量测序技术的发展和测序成本的持续降低,研究界和医学界不断产生海量的测序序列,然而传统方法从高通量序列中识别多肽效率低下。为了提高多肽的识别效率,基于机器学习的多肽识别方法越来越受到研究界的青睐[5]。近年来,研究界已提出了许多基于机器学习的治疗肽的预测模型,根据其使用算法进行分类,分为基于传统的机器学习肽预测模型与基于深度学习的肽预测模型。
基于传统的机器学习肽预测模型主要使用不同的序列特征把肽序列表示为特征向量,构造二分类样本集,使用不同的分类模型进行训练,然后预测新的肽序列。主要工作如下:2007年7月,Lata等人利用抗菌肽中N端和C端残基的特异性分别建立了基于神经网络、QM(quantitative matrices)和支持向量机的ABP预测模型[5];2017年5月,Wei等人整合了基于序列的特征描述符PC-PseAAC(parallel correlation pseudo-amino-acid composition)、SC-PseAAC(series correlation pseudo-amino-acid composition)、ASDC(adaptive skip dipeptide composition)、PPs(physicochemical properties),构建了基于随机森林算法的两层CPP预测框架CPPred-RF[6];2017年7月,Li等人使用OAAC(optimized amino acid composition)和ODPC(optimized dipeptide composition)两种特征开发了基于支持向量机的SBP预测器PSBinder,它可以快速有效地排除假阳性肽,更准确地获得SBP[7];2018年6月,Wei等人提出了一个基于支持向量机的ACP预测器ACPred-FL[8],使用了BPF(binary profile features)、GDC(G-gap dipeptide composition)、OPF(overlapping property features)、CTD(composition-transition-distribution)4种序列特征表示样本,通过最大相关-最小冗余和顺序前向搜索特征选择方法剔除冗余特征,提高了预测器的预测性能。以上预测方法都是针对识别特定的肽而构造的模型,2019年4月,Wei等人提出了基于随机森林的多肽预测模型PEPred-Suit,该模型引入了一种自适应特征表示策略,可以学习不同肽类型的最具代表性的特征,能有效识别多种不同类型肽[9]。
深度学习主要使用卷积神经网络和循环神经网络自动抽取出抽象特征,其中循环神经网络主要用于处理文本和序列数据。肽是一种序列数据,因此循环神经网络更适合肽的预测研究。针对基于深度学习的肽预测模型,2019年9月,Yi等人使用两种序列特征K-mer稀疏矩阵和BPF(binary profile features),构建了基于长期短期记忆LSTM(long short-term memory)循环神经网络的ACP预测模型ACP-DL,实现了一个DeepLSTM模型来自动学习如何识别抗癌肽和非抗癌肽。在基准数据集五折交叉验证实验结果表明,ACP-DL具有较高的识别性能[10]。
已有的基于机器学习肽预测方法促进了肽的研究,但分类器的识别性能仍有待提高,而且除了PEPred-Suit模型外,其他模型都只能识别某一种特定肽。针对以上问题,该文提出了一种通用的基于GRU循环神经网络的多肽预测模型DeepPEPred,能有效识别多种类型的肽。DeepPEPred用如下四种特征作为输入序列的编码:氨基酸组成(amino acid composition,AAC)、K-spaced氨基酸对的组成(composition of k-spaced amino acid pairs,CKSAAP)、构成/变迁/分布(composition/transition/distribution,CTD)、伪氨基酸组成(pseudo-amino acid composition,PAAC)能够有效预测不同的肽段,其中AAC在ACPred-FL模型被使用预测抗癌肽,CTD在PEPred-Suit模型被使用预测各种类型的肽。通过初步的实验验证:这四种特征使得DeepPEPred模型能够获得较好的总体性能。为了验证DeepPEPred的性能,该研究在抗癌肽、抗菌肽、细胞穿透肽和结合肽四种不同肽数据集上进行实验。经过十折交叉验证和独立测试结果表明,与现有的肽预测模型相比,DeepPEPred模型具有更强的识别性能。
1 数据集
该文旨在构建一个通用的深度学习模型预测具有不同治疗特性的肽,使用ACP、ABP、CPP和SBP四种肽数据集评估提出的模型,每种肽数据集包括一个训练集和一个独立测试集,训练集和独立测试集都由正例样本和负例样本组成,正例样本是经过实验验证的治疗性多肽(如抗癌活性),负例样本通常是没有相关特性(如非抗癌活性)或随机序列的多肽[9]。
该研究使用的ACP数据包括文献[9-13]提供的数据集和数据库CancerPPD[14]中最新的ACP数据,为了避免整合后序列中含有重复序列,该研究使用CD-HIT软件[15]去除同源性超过90%的序列。最后得到的ACP训练集中包括422个经实验验证的ACP序列以及1 688个非ACP序列;ACP独立测试集中包括97个经实验验证的ACP序列以及97个非ACP序列。该文使用了Lata等人[5]提供的ABP数据集、Wei等人[6]提供的CPP数据集以及Li等人[7]提供的SBP数据集。四种肽数据集的详细信息如表1所示。

表1 四种肽数据集
2 特征提取
该研究通过iLearn[16]选取了四种特征表示肽序列,分别是:氨基酸组成(AAC)、K-spaced氨基酸对的组成(CKSAAP)、构成/变迁/分布(CTD)、伪氨基酸组成(PAAC)。
2.1 氨基酸组成(AAC)
氨基酸组成(AAC)[17]是计算肽序列中每种氨基酸的出现频率,AAC特征编码的维度为20,序列中每种氨基酸出现的频率可由公式(1)计算:

(1)
其中,R(i)是肽序列中名称为i的氨基酸出现的次数,L是肽序列的长度。最终可以得到20种氨基酸在肽序列中的出现频率。
这个蜘蛛精……是人?青辰一边听着天葬师的话,一边仔细打量。那唐飞霄矮小瘦弱,整个身子都裹在硬甲中,只有一颗硕大的光头露在外面,看起来怪诞而不合比例。自己先入为主,竟将其当做了蜘蛛精,着实闹了个笑话。
2.2 K-spaced氨基酸对的组成(CKSAAP)

2.3 构成/过渡/分布(CTD)
CTD使用组成(C)、过渡(T)和分布(D)三个描述符描述蛋白质序列中[18]的每个基团中各性质的氨基酸分布,CTD采用七种物理化学性质表示蛋白质或肽序列,它们包括疏水性、标准化范德华体积、极性、极化度、电荷、二级结构和溶剂可及性,ilearn包[16]中将疏水性又分为七个不同性质,加上其他六种性质,共有13种性质。基于主要的氨基酸指数,针对每一种性质,将20种氨基酸分为三类。本研究只使用描述符D来编码肽序列,D统计三类氨基酸中每类氨基酸含量为0%,25%,50%,75%,100%时相对于整条肽序列的分布情况,即每类有五个描述符值,因此每种性质使用3×5=15个描述符表示。因此,CTD将一个肽序列编码成一个由13×15=195个描述符值组成的向量。
2.4 伪氨基酸组成(PAAC)
传统的氨基酸组成只考虑蛋白质序列中20个氨基酸出现的频率,这会丢失蛋白质链的序列信息。PAAC将20个氨基酸的序列顺序信息和频率整合在一起进行编码[19]。一个蛋白质序列编码成一个20+A维向量,向量的前20个分量表示20个氨基酸的出现频率,最后的A个分量表示序列顺序信息。PAAC被证明是一种有效的特征编码方案,并被广泛应用于蛋白质序列或者肽序列相关领域的研究[20]。输入肽序列的PAAC计算由ilearn包提供。经实验验证,当A=4时,模型预测性能最优,因此PAAC将一个肽序列编码成一个24维特征向量。
3 特征标椎化
不同特征向量往往具有不同的量度,这将影响到模型预测性能,因此需要对原始特征组合进行标准化使得每个特征处于同一数量级,有利于预测模型的建立[21]。
该研究使用的Z-score方法是基于原始特征的均值(mean)和标准差(standard deviation)进行数据的标准化,该方法适用于数据属性值的最大值和最小值未知的情况,或有超出取值范围的离群数据的情况[21]。Z-score标准化可由公式(2)计算:

(2)
其中,Z为标准化后的变量值,X为实际变量值,μ为各变量(特征)的算术平均值(数学期望),σ为标准差。
4 基于深度学习的肽识别方法
深度学习(deep learning,DL)作为机器学习的新兴技术[22],近年来已被广泛应用于生物信息学中[23]。深度学习模型包括卷积神经网络模型、堆栈自编码网络模型、长短期记忆网络模型(long short-term memory,LSTM)[24-25]等。
4.1 模型的整体框架
提出的基于深度学习的多肽识框架如图1所示,主要包含以下几个步骤。

图1 DeepPEPred预测方法流程
步骤一:肽序列数据集构造。该研究收集了ACP、ABP、CPP和SBP四种肽数据集,四种数据集分别包含一个训练集和一个独立测试集,每种数据的训练集和独立测试集见表1。
步骤二:肽序列样本集表示。该研究通过对多种肽序列特征进行性能评估,筛选出四种对于ACP、ABP、CPP、SBP序列有较强识别能力的特征,四种特征分别是AAC、CKSAAP、CTDD、PAAC,它们编码维数分别是20、2 000、195、24,每一个肽序列样本被编码的维度为2 239,得到四种肽序列的二分类样本集。
步骤三:深度学习模型构建。输入层特征维度为2 239个,将输入层神经元输入隐藏层,隐藏层的第一层为GRU层,输出维度为59,GRU层后面增加一个Dropout层,设置为0.465,防止模型过拟合;输出层空间维度为1,使用sigmoid激活函数。在模型训练过程中,使用early-stop早停机制,防止模型过拟合;损失函数使用交叉熵损失函数,优化器使用Adam,迭代次数(epoch)为100次。
步骤四:模型训练。该研究先使用ACP数据集训练一个初步的预测模型,由于ACP数据集中负例样本数是正例样本数的四倍,样本集严重不平衡,将影响模型的性能。该研究借鉴BootStrapping[28]方法来解决数据集中正负例样本不平衡问题,BootStrapping方法是指对数据集进行有放回的抽样,将每次抽取的数据作为一个新样本,重复多次,形成多个新样本。该研究对负例样本集采取不放回抽样方法,该策略的示意图如图2所示。假设P和N分别表示正例样本集(ACP序列)和负例样本集(非ACP序列),TP和TN表示正例样本和负例样本的数量,以大小为TP的窗口循环遍历负例样本集,循环n=TN/TP次,每次循环抽取的TP个负例样本作为一个负子集,与正例样本集结合生成一个正负例数目相同训练集,并用这个训练集进行模型训练,保留每次循环训练的模型,最终预测结果取n次模型预测结果的均值。

图2 ACP数据集划分图
步骤五:模型优化。首先在ACP数据集上经过贝叶斯参数调优[29]进行参数寻优,得到一个最优参数的框架,然后用最优参数框架训练ABP、CPP、SBP数据集。贝叶斯优化方法首先采用高斯过程不断地更新目标函数的后验分布,然后在预先设置的参数范围内自动搜索最好的参数。在参数优化时,设置GRU层输出维度的初始范围为[8,128],优化后的最优值为59;设置Dropout的初始范围为[0.1,0.6],优化后的最优值为0.465。经过上述操作确定了最优参数,并构建了一个适用于四种治疗肽的最优模型。
步骤六:模型评估。该研究使用十折交叉验证和独立测试方法对模型进行评估,并与现有模型进行预测性能比较。
4.2 评价指标
为了评估DeepPEPred模型的预测性能,该研究使用了五种常用指标来评价模型的性能,包括AUC(area under the ROC curve)值、准确度(accuracy,Acc)、特异性(specificity,Sp)、敏感性(sensitivity,Sn) 和马修斯相关系数(Matthews correlation coefficient,MCC)。其中AUC表示ROC(receiver operating characteristic)曲线下的面积,ROC曲线是指按顺序逐个对样本进行预测,每次计算出真阳性率(TPR)与假阳性率(FPR)分别以它们作为纵、横坐标进行绘制而生成的曲线。较大的AUC值表示该模型实现了更好和更强大的预测性能。这五种评价指标的定义如下:
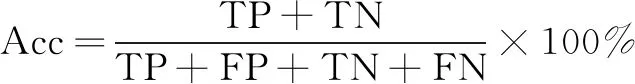
(3)
(4)
(5)
MCC=
(6)
其中,TP、TN、FP和FN分别表示真阳性、真阴性、假阳性和假阴性的样本数量。
5 实验结果
该研究对比的ACP识别方法在同样的数据集上采用独立测试,其他三种肽的识别方法在相同的数据集上采用十折交叉验证方法,为了公平比较,该研究分别采用同样的策略。
5.1 十折交叉验证
图3表示DeepPEPred与现有模型在ABP、CPP、SBP三种肽数据集上十折交叉验证结果的比较。由于现有的模型仅仅通过AUC值进行评价,为了公平对比,该研究也仅仅提供了每种数据集的AUC值。

图3 DeepPEPred和现有预测器在ABP、CPP和
从图3的结果可知:在相同数据集上DeepPEPred预测模型在AUC方面取得了比其他预测方法更好的性能。在三个数据集(ABP、CPP和SBP)上比目前最新模型PEPred-Suite的AUC值分别高0.8%、0.3%和1.2%,比其他预测同类型肽模型(AntiBP、CPPred-RF和PSBinder)的AUC值分别高出2.7%、1.3%和5.9%。
在表2分别给出了DeepPEPred和PEPred-Suite在三个数据集上十折交叉验证的其余指标(Acc、Sn、Sp、MCC)的值,在ABP数据集上实验结果表明:DeepPEPred比PEPred-Suite的MCC和Acc分别高出2.3%和1.2%;在CPP数据集上实验结果表明:DeepPEPred比PEPred-Suite模型的MCC和Acc分别高出2.5%和1.2%;在SBP数据集上实验结果表明:DeepPEPred比PEPred-Suite模型的MCC和Acc分别高出2.4%和1.2%。

表2 ABP、CPP、SBP数据集上十折交叉验证结果
5.2 独立测试
为了验证DeepPEPred的泛化能力,该研究在ABP、CPP和SBP数据集上进行了独立测试,并与现有方法进行了性能比较,结果如图4所示。从图4中结果可知:在三个数据集上与PEPred-Suite预测模型相比,AUC值分别提升了0.7%、1.5%和1.0%。在ABP数据集上,DeepPEPred与同类型肽预测模型AntiBP相比AUC值分别提升了0.7%;在CPP数据集上,DeepPEPred与在同类型肽预测模型CPPred-RF相比AUC值提升了2.6%;在SBP数据集上,DeepPEPred与PSBinder的AUC值相等。
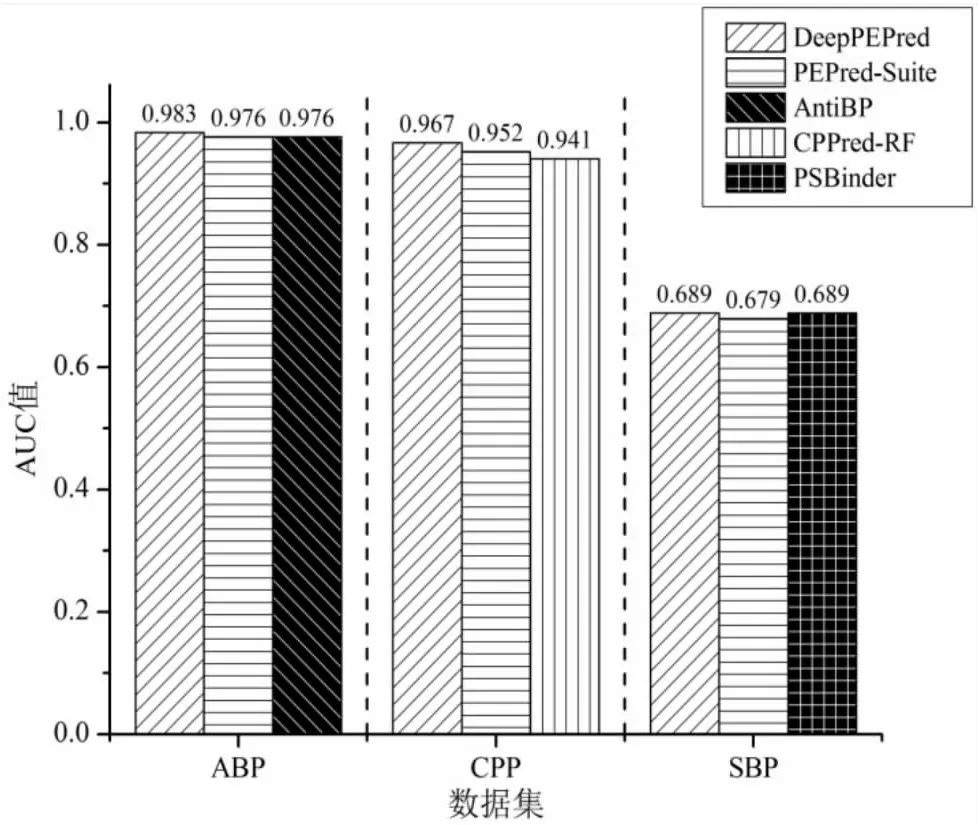
图4 DeepPEPred和现有预测器独立测试的性能对比
DeepPEPred模型在ABP、CPP和SBP数据集上独立测试的MCC、Acc、Sn、Sp评价结果如表3所示。即使DeepPEPred是基于ACP数据集构建及调优的,从图4和表3结果可知,DeepPEPred对于ABP、CPP和SBP三种肽预测也是有效的,说明DeepPEPred具有较强的迁移能力。

表3 ABP、CPP、SBP独立测试详细结果
由于ACP数据集正负例样本不平衡,该研究首先将负例样本分成四份,每份与正例样本相结合生成四个正负均衡的训练集;然后对模型进行四次训练,每次训练得到的模型进行独立测试,测试结果如表4所示,最终结果为四次结果的均值,其AUC、MCC和Acc最终值分别为0.875、0.631和0.811。

表4 ACP数据集独立测试结果
为了进一步验证DeepPEPred模型预测ACP的性能,该研究比较了DeepPEPred与PEPred-Suite、ACPred[30]两个ACP预测模型,独立测试结果如表5所示。需要强调的是,PEPred-Suite和ACPred独立测试结果是使用对应文献中提供的在线预测平台测试获得的。从表5的结果可知:DeepPEPred相对于PEPred-Suite和ACPred,在Acc、MCC、Sp值方面都有较为显著的提升,其中Acc值分别提升了29.6%和4.3%,MCC值分别提升了59.7%和9.4%,Sp分别提升了17.5%和10.3%,Sn相比PEPred-Suite提升了41.5%。这说明了该研究提出的模型对于ACP预测是有效的。

表5 不同模型预测ACP的性能对比
6 结束语
提出了一种基于深度学习的多肽预测方法DeepPEPred。该方法利用四种特征对输入序列进行编码,将标准化的编码作为模型输入,经过贝叶斯参数调优,构建出一个最优的多种肽预测模型。
该方法的主要贡献是构造一个通用的模型,能有效预测多种肽。DeepPEPred模型对不同的多肽表现出一致的鲁棒性,说明它具有很强的泛化能力。在四种肽数据集上与现有的方法进行了对比,实验结果表明:DeepPEPred模型在AUC、Acc和MCC三个综合性评价指标上比现有的预测方法更好。
猜你喜欢
数学年刊A辑(中文版)(2020年2期)2020-07-25 02:04:44
数学物理学报(2019年6期)2020-01-13 06:08:16
中国洗涤用品工业(2019年4期)2019-05-11 09:27:18
中成药(2018年1期)2018-02-02 07:20:05
现代园艺(2017年13期)2018-01-19 02:28:09
数学物理学报(2017年5期)2017-11-23 07:51:31
现代检验医学杂志(2016年3期)2016-11-15 01:59:28
动物医学进展(2015年10期)2015-12-07 05:46:19
药学与临床研究(2015年4期)2015-06-05 11:35:54
科学中国人(2015年16期)2015-02-28 09:14:02
