
高效液相色谱法测定血浆中甘草西定浓度
2021-07-31 02:40王牧原
锦州医科大学学报 2021年3期
王牧原
(锦州医科大学附属第三医院,辽宁 锦州 121000)
甘草在我国是一种古老的药用植物,甘草的根茎具有显著的抗病毒、抗炎、抗癌、抗过敏、保肝和雌激素活性[1-3]。甘草西定是甘草的主要成分之一,其具有有很强的抗菌[4-5]、抗癌[6]、抗炎[7]活性。此外,甘草西定可防止UVA诱导的人皮肤成纤维细胞的光老化[8]。在研究甘草西定药代动力学特征之前,需要测定甘草西定的血药浓度,然而甘草西定的血浆浓度测定至今没有相关报道。高效液相色谱法(high performance liquid chromatography,HPLC)是一种重要的检测方法[9-11],其能够定量检测药物在血浆中的浓度。本研究利用HPLC对甘草西定血浆浓度进行测定,并对方法学进行了验证,以期为今后甘草西定药代动力学研究提供分析方法,见图1。
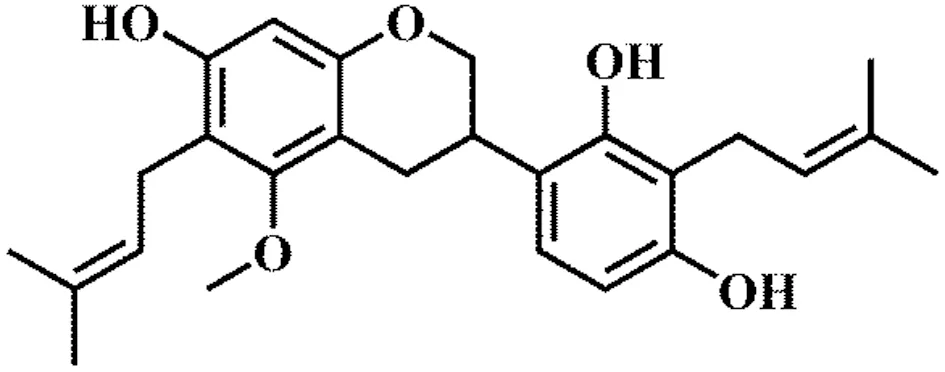
图1 甘草西定结构式
1 仪器与材料
1.1 仪器
日本岛津 LC-20AB高效液相色谱仪,其中包括CTO-20AB柱温箱,SIL-20A自动进样器和 SPD-20A紫外检测器;UV752紫外可见分光光度计(上海诺科);贝克曼高速离心机(美国Beckman Coulter公司);XB220A万分之一电子天平(瑞士Precisa公司);HERMO.SHAKER金属混匀仪(英国Grant仪器有限公司);Synergy Uv纯水仪(美国Millipore公司),氮吹仪(CM-12,北京成萌伟业科技有限公司),可调移液器(eppendorf,德国)等。
1.2 试剂
甘草西定对照品由云南西力生物技术股份有限公司提供,纯度98%;乙腈和甲酸都是色谱级(德国默克);其他试剂为分析纯,水为纯化水。
2 方法与结果
2.1 标准溶液的配制
标准贮备液的配制:精密称取甘草西定对照品2 mg,置10 mL量瓶中,用乙腈定容至刻度,得浓度为0.2 mg/mL的贮备液,置4 ℃冰箱保存,临用时稀释至所需浓度。
标准工作液的配制:将甘草西定标准贮备液用乙腈稀释定容,配制成质量浓度分别为100、50、10、5、1、0.5、0.1、0.05、0.01 μg/mL 的甘草西定标准工作液。
2.2 测定波长的选择
配制10 μg/mL甘草西定溶液2 mL,用乙腈作为对照液,在200~400 nm波长范围内扫描,甘草西定在207 nm 和 285 nm处有等吸收,为了降低溶剂效应我们选择285 nm作为甘草西定的测定波长。
2.3 色谱条件
采用Hypersil BDS C18反相分析色谱柱(大连依利特分析仪器有限公司,5 μm,4.6 mm×150 mm),柱温设置40 ℃,流动相由0.1%的甲酸水和乙腈组成梯度洗脱。流速为 1 mL/min,检测波长为285 nm,进样体积10 μL,见表1。
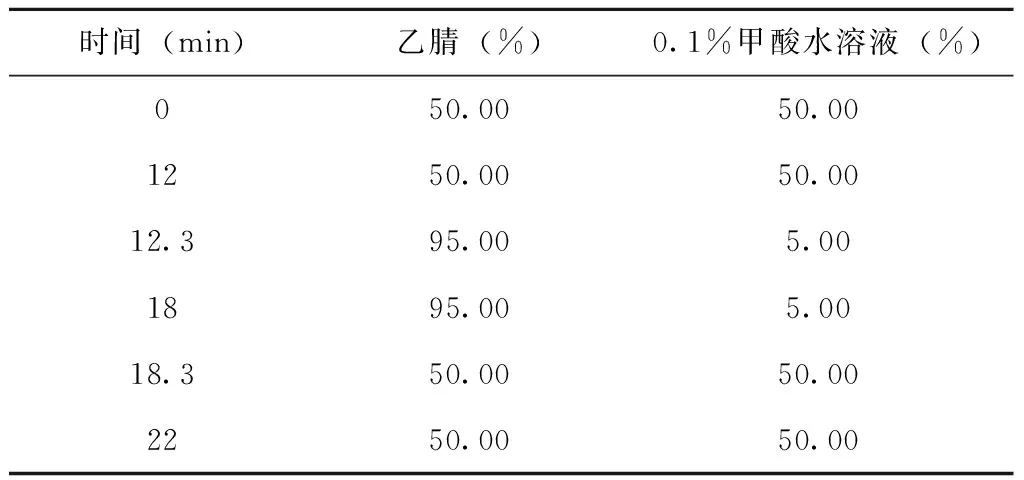
表1 甘草西定分析的梯度洗脱程序
2.4 血液样品处理
取血浆0.3 mL,加10 μL甘草西定贮备液(0.2 mg/mL),混匀后加乙酸乙酯1 mL,振摇3 min后离心(10 000 r/min,15 min),取出有机相,用氮气流吹干,100 μL 乙腈溶解残渣,10 μL进样分析。
2.5 方法的专属性试验
试验分为3组,50 ng/μL标准液直接进样组、空白血浆组和含甘草西定血浆组(50 ng/μL)。空白血浆组和含甘草西定血浆组按“2.3”方法进行血浆样品处理。3组在“2.2”项色谱条件下,分别直接进样。甘草西定保留时间为15.34 min,杂质没有干扰,见图2。
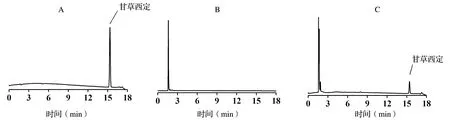
A:甘草西定对照品溶液;B:空白血浆;C:含甘草西定血浆
2.6 标准曲线的制备
取空白血浆0.5 mL,精密加入一定量的甘草西定标准溶液,使血浆中甘草西定的浓度分别为 0.1、0.3、0.5、1、3、10 μg/mL。并按“2.4”项下方法操作,记录色谱。分别以甘草西定的浓度(X)为横坐标,峰面积(Y)为纵坐标作直线回归,得直线回归方程分别为:Y=26865X-8199,R2=0.9993。结果表明,甘草西定检测浓度在0.1~10 μg/mL范围内与其峰面积线性关系良好。按信噪比S/N=1/2计算,最低检测限为0.05 μg/mL。
2.7 精密度和回收率
配制浓度为0.1、1、10 μg/mL的甘草西定标准血浆样品各5份,按“2.4”项下处理,于同一天内测定血样中甘草西定的浓度,求得该方法的日内精密度,另将测定值除以理论值即为本测定方法的相对回收率,血浆测定的峰面积除以相同浓度化学样品直接进样的峰面积比值即为样品的绝对回收率。同法配制甘草西定高、中、低的样品各5份,置冰箱冷冻,自配制之日起,每日取出1份测定甘草西定的浓度,计算每种浓度样品的 RSD 值,求得日间精密度。精密度及回收率试验结果,日内RSD < 10%,日间精密度<15%,见表2。
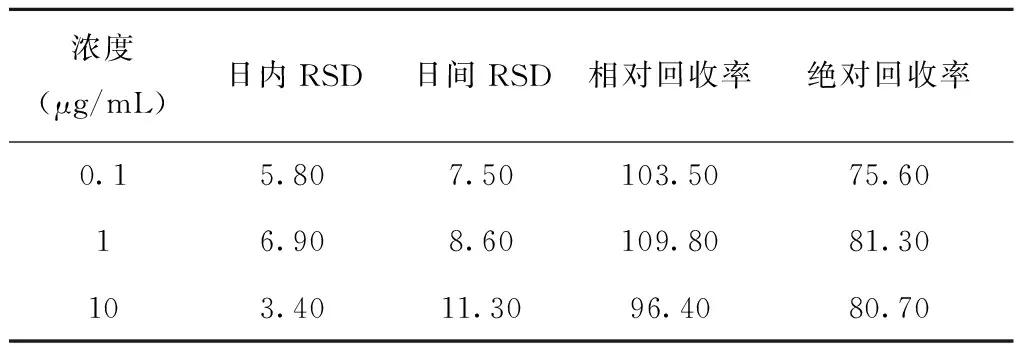
表2 精密度及回收率试验结果(%)
2.8 乙腈中甘草西定在室温的稳定性试验
将0.1、1、10 μg/mL甘草西定标准品溶液在室温下放置第0、8、16、24小时后,按“2.3”色谱条件用高效液相色谱测定其浓度,进样量为10 μL。显示甘草西定乙腈溶液在室温下24小时内稳定性较好,见表3。
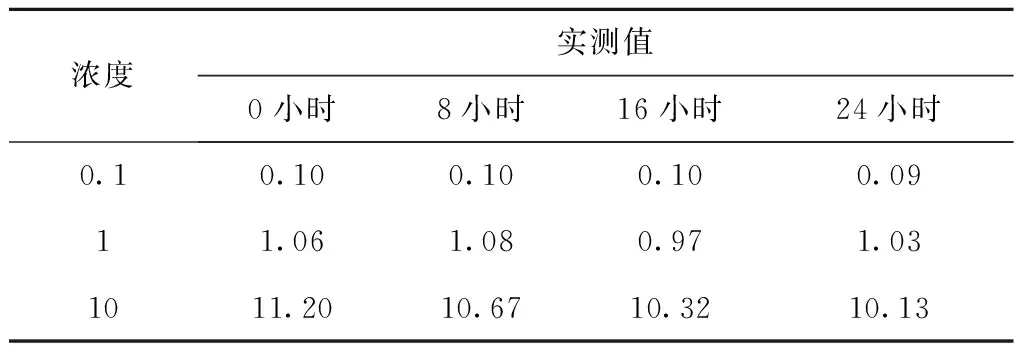
表3 不同时间乙腈中甘草西定的浓度(μg/mL)
2.8 甘草西定工作液在低温条件长期保存的稳定性试验
配制0.1、1、10 μg/mL甘草西定工作液,放入-20 ℃冰箱中冷冻,分别存放0、1、2和4个月后取出测其浓度,考察甘草西定在长期保存过程中的稳定性。结果显示长期保存并不影响甘草西定的稳定性,见表4。
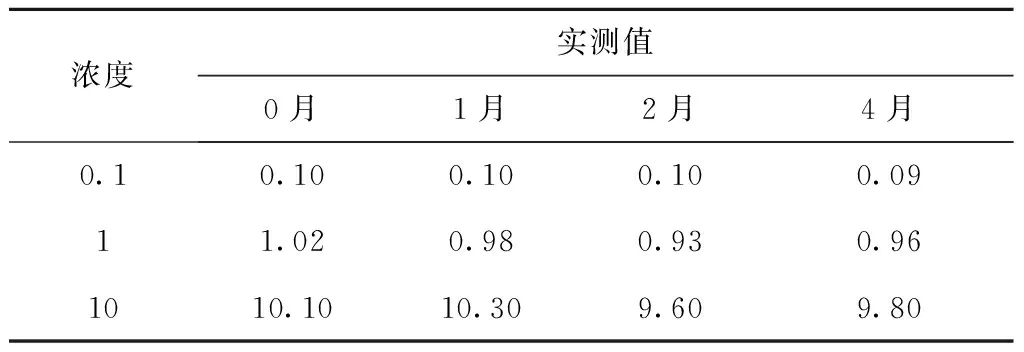
表4 甘草西定在乙腈中冰冻稳定性(μg/mL)
2.9 血浆中甘草西定多次冻融稳定性试验
在空白血浆中加入适当甘草西定工作液,配制成0.1、1、10 μg/mL的甘草西定血浆样品20 mL,然后各取0.5 mL分装于离心管中,放入-20 ℃冰箱中冷冻,取出室温解冻,连续冻融3次,每次按照血浆样品处理后测含量,考察样品冻融稳定性。结果显示反复冻融3次并不改变血药浓度,见表5。
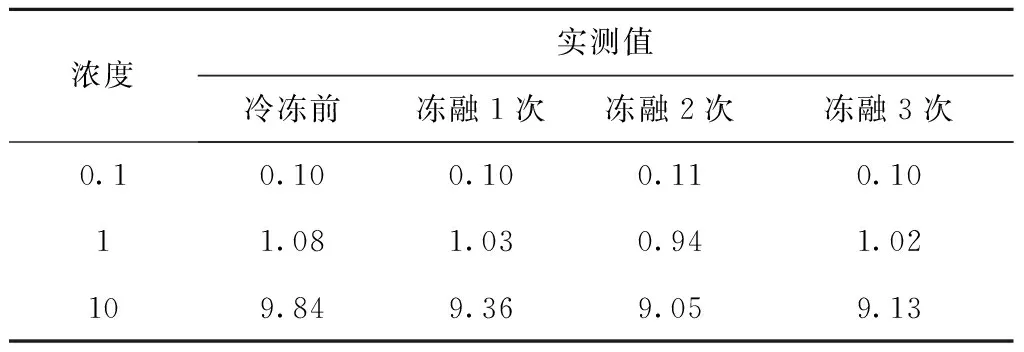
表5 冻融试验结果(μg/mL)
2.10 甘草西定血浆中长期保存稳定性试验
在空白血浆中加入适当甘草西定工作液,配制成0.1、1、10 μg/mL的甘草西定血浆样品20 mL,然后各取0.5 mL分装于离心管中,放入-20 ℃冰箱中冷冻,分别在第0、1、2和第4周取出,每次按照血浆样品处理后测含量,考察样品在长期保存过程中的稳定性。结果显示长期保存并不影响血药浓度,见表6。
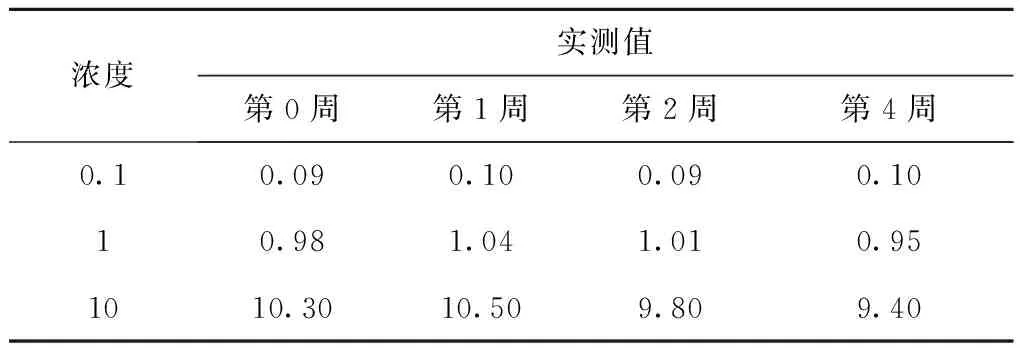
表6 甘草西定在血浆中的冰冻稳定性(μg/mL)
3 讨 论
血药浓度的检测在药物的研发和药物的应用都具有重要的意义,如在药物的研发阶段通过血药浓度来获知药代动力学参数从而为药物的使用提供依据,在药物的应用阶段通过测定药物在血浆中的浓度使药物在达到治疗效果同时避免不良反应的发生。常见的血药浓度检测的方法包括分光光度法、气相色谱法、高效液相色谱法和免疫学方法。而高效液相方法是分析速度最快,应用范围最广的血药浓度检测方法[12-13]。
有关甘草西定血药浓度的测定方法目前没有相关文献报道,本实验采用HPLC 紫外检测方法,首先用紫外分光光度计扫描甘草西定标准品,测得最大吸收波长为285 nm。因为甘草西定在乙腈中溶解性较好,因此流动相选用乙腈和0.1%甲酸水。在流动相中加入甲酸是因为甘草西定结构中有3个酚羟基,加入甲酸能使甘草西定在流动相和色谱柱中以分子形式存在,峰型更好。另外,我们曾采用梯度洗脱,流动相是由乙腈和0.1%甲酸水组成,两者的极性不同,通过改变两者在流动相中的比例从而改变流动相的极性,各组分在色谱柱中都有较好的容量因子k,并使甘草西定和其他组分在较短时间内达到较好的分离效果,甘草西定的保留时间合适。
由于生物样品的组成复杂,基质对待测物的干扰较严重,并且待测物在样品中的含量较低。因此,如何能从复杂的生物样品中快速、准确地提取和分离目标成分,并达到分析仪器的检测标准,需要对生物样品进行预处理。生物样品的处理方法包括沉淀蛋白和有机溶剂萃取等方法[14-15]。沉淀蛋白方法操作步骤简单,向血清或血浆中加入一定量的有机溶剂、无机盐或酸性物质等,样品中的蛋白质遇到这些物质会发生变性反应,导致蛋白沉淀,沉淀的蛋白质经高速离心去除。蛋白沉淀法特别适用于强极性药物或两性类药物,这些药物难以用有机溶剂从血浆中提取。由于乙腈和甲醇对液相色谱分析和液相色谱-质谱的兼容性好,所以最常用的沉淀蛋白的有机溶剂为乙腈和甲醇[16]。我们先试用高氯酸和乙腈等沉淀蛋白,离心后取上清液直接进样的方法,结果表明回收率和检测限不能满足要求且在甘草西定出峰时间有杂质干扰。因而采用液液萃取方法从血浆中提取样品,通过氮吹富集后回收率和检测限可满足测定要求,样品峰没有干扰。
综上所述,本文首次建立了血浆中甘草西定的高效液相测定方法,该方法精密度高,重现性好,回收率符合要求,方法科学,结果准确,易于推广。
猜你喜欢
四川生理科学杂志(2022年9期)2022-10-10
煤化工(2022年3期)2022-07-08
中国现代医生(2022年6期)2022-04-23
中山大学学报(自然科学版)(中英文)(2022年2期)2022-04-12
药品评价(2021年22期)2022-01-09
中老年保健(2021年9期)2021-08-24
家庭百事通·健康一点通(2020年8期)2020-09-08
心理与健康(2020年3期)2020-03-25
中国信息化·学术版(2013年3期)2013-06-25
