
Bacterial community shifts in diff erent functional zones of a reservoir with ecological purification facilities*
2021-07-29 09:11LeiZHANGYueZHANJiahuJIANGWenxuanLU
Lei ZHANG , , Yue ZHAN , Jiahu JIANG , Wenxuan LU
1 School of Civil Engineering and Architecture, Chuzhou University, Chuzhou 239000, China
2 State Key Laboratory of Lake Science and Environment, Nanjing Institute of Geography and Limnology, Chinese Academy of Sciences, Nanjing 210008, China
3 Fisheries Research Institute, Anhui Academy of Agricultural Sciences, Hefei 230001, China
Abstract Ecological purification in a reservoir is an important strategy to control the level of nutrients in water. The bacterial community of such a reservoir is the main agent for pollutant degradation, which has not been fully documented. Taking the Jinze Reservoir, a freshwater source for Shanghai, China as the case, its spatial distributions of water and sediment bacteria were determined using 16S rRNA gene-based Illumina MiSeq sequencing , and the environmental parameters were analyzed. The reservoir takes natural river water and functions as an ecological purification system, consisting of three functional zones, i.e., pretreatment zone, ecological purification zone, and ecological sustaining zone. Results show that the concentrations of both total nitrogen (TN) and total phosphorus (TP) decreased considerably after the ecological treatment,and the concentration of dissolved oxygen (DO) in the ecological purification zone was boosted from that before pretreatment. In addition, patterns of bacterial communities in both water and sediment were similar and consisted of mainly Proteobacteria, Actinobacteria, and Bacteroidetes. However, diff erence in water bacterial composition was distinct in each functional zone, whereas the bacterial communities in sediment changed only slightly among sediment samples. Network analysis revealed nonrandom co-occurrence patterns of bacterial community composition in water and sediment, and Pseudomonas, unclassified Comamonadaceae, Variovorax, and Dechloromonas were the key taxa in the co-occurrence network. The bacterial taxa from the same module of the network had strong ecological connections, participated in C-cycles, and shared common trophic properties. PICRUSt analysis showed that bacteria were involved potentially in various essential processes; and the abundance of predicted xenobiotic biodegradation genes showed a decreasing trend in water samples from the inlet to the outlet of the reservoir. These results improve our current knowledge of the spatial distribution of bacteria in water and sediment in ecological purification reservoirs.
Keyword: ecological purification reservoir; bacterial community; Illumina MiSeq sequencing; network analysis; PICRUSt analysis
1 INTRODUCTION
Nutrient enrichment in reservoirs freshwaters is a serious ecological and economic issue in the world(Soto and Mena, 1999). High nutrient concentrations in reservoirs can cause diffi culties for water purification and supply (Parsons and Jeff erson, 2006).In recent years, countermeasures to reservoir eutrophication include permeable substrata (gravel)settlement and aquatic flora planting (Typha,Phragmites, and Cyperus), among others (Haberl et al., 2003); and related research has shown that planting aquatic flora has positive eff ects on the water restoration (Zhu and Sikora, 1995). The water quality in Wuli Lake (Jin et al., 2006) and Taihu Lake in China (Qin et al., 2006), Norwegian Lake (Norway)(Rooney and Kalff , 2003), and Massaciuccoli Lake(Italy) (Ciurli et al., 2009) has been significantly improved by implementing the scheme. As a result,some countries have begun to build an ecological purification system in reservoir in recent years (Ding et al., 2018; Ye, 2018).
Ecological purification reservoirs are an organic system integrating ecological purification technology,ecological restoration technology, and physical algaecontrol technology, and are designed to construct a water quality restoration system for the reservoir water (Ding et al., 2018), in which polluted water is purified through the interaction of aquatic plants and bacteria, achieving physical, and chemical eff ect(Lauridsen et al., 1996; Körner and Nicklisch, 2002;Xie et al., 2009). Aquatic plants play an important role in water purification, and the plants can directly absorb nutrients in water. In addition, through a series of mechanisms with physicochemical functions, bacteria in water and sediment can adsorb and decompose the pollutant (Zhao et al., 2008) and thus achieve eff ective removal of nitrogen, phosphorus, and various organic substances in water (Whitacre, 2009).
Bacteria are a crucial part of food web and they aff ect biogeochemical cycles in water ecosystems(Sarmento and Gasol, 2012) in the courses of organic decomposition and nutrient recycling (Fernandez et al., 2016). Given that bacterial communities play a key role and their rapid response to the changes in ecosystems (de Figueiredo et al., 2007), the abundance, community composition, diversity, and interaction (e.g., competition, mutualism, and antagonism) can be used to assess the eff ects of ecological restoration in reservoirs with ecological purification facilities (Deng et al., 2012). However,due to diff erences in bacterial community habitat in diff erent functional zones of the reservoirs and in the composition and the spatial distribution of bacteria communities in water and sediment, the driving forces of bacterial assemblages remain elusive (Ye et al.,2009). Thus, to assess the comprehensive ecological impact, it is important to investigate the bacterial community composition in water and sediments in reservoirs with ecological purification ability.
Current analytic methods (such as high-throughput sequencing) analyze the composition and diversity of bacterial communities only and do not provide an indepth understanding of complicated bacterial communities and the interactions among bacteria.Thus, it is diffi cult to obtain a comprehensive community profile in complex environments (Sun et al., 2014). Bacteria live in complex networks via a multitude ofinteractions (e.g., competition,mutualism, and antagonism) (Deng et al., 2012).However, most of the interactions cannot be directly observed. Co-occurrence network analysis has recently been used to provide important information beyond sample-level comparisons (Deng et al., 2012;Faust and Raes, 2012; Banerjee et al., 2016b).Therefore, the construction of co-occurrence networks can be used to study bacterial communities in more detail. However, the preliminary exploration of a cooccurrence model does not establish a strong relationship between bacteria (Jiao et al., 2016). We introduced PICRUSt—a bioinformatics software package—for providing potentially new insights into the changes in ecosystem functions under diff erent environmental conditions (Roberto et al., 2018), thus achieving the prediction of bacterial metabolic functions (Verma et al., 2017). However, the results must be interpreted carefully as PICRUSt cannot prevent or outperform deep metagenomic sequencing(Langille et al., 2013). Therefore, deeper sequencing is required for full assessment of gene categories.
Therefore, we used 16S rRNA gene Illumina MiSeq sequencing to study the composition and diversity of bacteria in water and sediment in the reservoir of ecological purification. Patterns ofinteractions in complex assemblages of bacteria were quantified via co-occurrence networks, and PICRUSt was applied based on the functional contributions of bacteria in ecological environments. In a broad scientific scope, the aim of this study was to explore the spatial changes of physical and chemical parameters of each functional area in a reservoir of ecological purification function, to study the spatial changes in bacteria in water and sediment in diff erent functional zones, and to elucidate the underlying mechanisms of co-occurrence of bacterial communities in water and sediment in ecosystems of the reservoir with ecological purification facilities.
2 MATERIAL AND METHOD
2.1 Study site and sampling
Jinze Reservoir (31°01′56″N, 120°57′21″E) is located in Jinze town, Shanghai, China, on the northern flank of the Taipu River (Ye, 2018). It is the water supply source for five southwestern districts of Shanghai (Qingpu, Songjiang, Jinshan, Minhang, and Fengxian, for approximately 6.7 million in population). The reservoir covers an area of approximately 2.7 km2, of which water area is 1.92 km2; the average water depth is 3.3 m, and the total storage is approximately 9.10×106m3(Ye, 2018).The reservoir water is taken directly from the Taipu River via a water transfer pump station. The reservoir constitutes a purification system of three functional zones, i.e., pretreatment zone, ecological purification zone, and ecological sustaining zone. Limited by the small size, the reservoir has a large flow but a short hydraulic retention time. In the pretreatment zone,large particles ofinorganic suspended matter settled and removed naturally. In the ecological purification zone, submerged plants (Myriophyllumspicatum,Vallisnerianatans, and genusElodea) and emergent plants (reed, cattailTyphaangustifolia, andAcoruscalamus) were grown to remove nitrogen and phosphorus nutrients, and the coverage of macrophyte was 3.66%. In the ecological sustaining zone,ecological gravel beds were laid to form a porous microbial enrichment space to intercept and degrade organic matter in the water (Qian and Chen, 2016; Ye,2018).
To understand the changes of water quality parameters and bacterial community composition from the inlet to the outlet of the reservoir, five sampling sites were selected at the inlet (JZ1)(31°01′03″N, 120°56′54″E), pretreatment zone (JZ2)(31°01′23″N, 120°56′49″E), ecological purification zone (JZ3) (31°02′03″N, 120°56′58″E), ecological purification zone (JZ4) (31°01′59″N, 120°57′14″E),and ecological sustaining zone (JZ5) (31°01′45″N,120°57′51″E) (Fig.1). Five composite water samples(0.5 m below water surface) (Xu et al., 2018; Mao et al., 2019) and five composite sediment samples were collected from the five sites in August 2018. The water samples were named JZW1, JZW2, JZW3,JZW4, and JZW5, and the corresponding sediment samples were JZS1, JZS2, JZS3, JZS4, and JZS5,respectively collected at each of the five sampling sites. At each sampling site, three parallel water samples were collected using a plexiglass water sampler (approximately 0.5-m sampling depth) and mixed into one sample, and the sediment was collected three times in parallel with a Petersen grab sampler and mixed. Water samples were stored in aseptic polyethylene bottles, and sediment samples were put into plastic bags. All the samples were transferred to laboratory in 10 h and stored at -20 °C for later analysis in the procedures as per Portillo et al. (2012)and Wang et al. (2018).
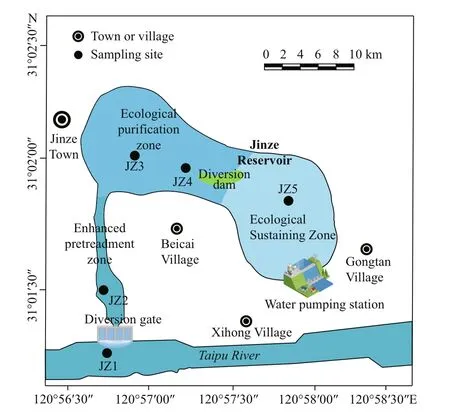
Fig.1 Schematic diagram of the sampling site in the Jinze Reservoir, Shanghai, China
2.2 Physicochemical analysis
The temperature (T), Chla, pH, and dissolved oxygen (DO) of water were measured by YSI sonde(YSI 6600V2 multi-parameter sonde, USA) after sample collection. According to the standard methods,the total nitrogen (TN), total phosphorus (TP), and total organic carbon (TOC) were measured in laboratory (Jin and Tu, 1990). All chemical measurements were conducted in triplicate.
2.3 DNA extraction and quality detection
According to the manufacturer’s instructions, the total water microbial nucleic acid (DNA) was extracted with 0.22-μm filters using a Power Water DNA Extraction kit (Mo Bio, CA, USA) and sediment of each sample (0.25 g) was taken to extract DNA(300-400 mL) after homogenization under aseptic conditions. The integrity of the DNA sample was verified in 0.8% agarose gel electrophoresis(Eppendorf, Germany), and quantified using an ultraviolet spectrophotometer. The DNA was then stored at -20 °C for further use.
2.4 PCR amplification and sequencing
The V4 region of the 16S rRNA gene was amplified using the primer set 515F/907R. The primer sets were 515F (5′-GTGCCAGCMGCCGCGGTAA-3′) and 907R (5-CCGTAACMTTTRAGTTT-3′). PCR amplification was performed in the procedure of 98 ℃ for 30 s, 27 cycles of 98 ℃ for 15 s, 50 ℃ for 30 s, 72 ℃ for 30 s, and 72 ℃ for 5 min. The PCR amplification products were detected using 2%Invitrogen agarose, and the target fragment was subject to gelation recovery using the AXYGEN gel recovery kit. The PCR amplification product was subjected to fluorescence quantification using a Quant-iT PicoGreen dsDNA Assay Kit and a Microplate reader (BioTek, FLx800). Afterwards,each sample was mixed in a corresponding ratio according to the sequencing amount required, and then the purified products were sent to Personal Biotechnology Co., Ltd., Shanghai, China for sequencing.
2.5 Sequencing data optimization and OTU clustering
The Illumina MiSeq system was used to perform paired-end sequencing of community DNA fragments.To minimize random sequencing errors and improve the read quality, FLASH was applied to double-ended sequences that were screened by mass were paired according to overlapping bases. With USEARCH software (Edgar, 2010) for the paired ends, the 16S readings were combined, primers and distal bases were trimmed, singletons were removed, and quality filtration was conducted. The RDP (version 2.2 http://sourceforge.net/projects/rdp-classifier/) classifier Bayesian algorithm (confidence threshold is 0.7) was applied to classify OTUs with 97% similarity to obtain species classification information corresponding to each OTU (Samarajeewa et al., 2015).
2.6 Data analysis
Principal coordinates analysis (PCoA) was used to perform beta diversity analysis based on the Bray-Curtis diff erence matrix (Mitter et al., 2017). To describe the correspondence relationship among species and samples (Sui et al., 2016), a visible circle diagram by Circos-0.67-7 (http://circos.ca/) software was used. The redundancy analysis (RDA) in R software (Oksanen et al., 2018) was also used to estimate the correlation between bacterial community composition and environmental variables. The collinear network model of the bacterial taxonomic group was built by selecting top 50 taxa in abundance,and the igraph package under the illumine adapter(Csárdi and Nepusz, 2006) was applied to calculate the number of nodes, the number of edges, and the minimum path length. In addition, the functional genes of the bacterial community and the metabolic potential were predicted with PICRUSt (Dong et al.,2018). An online analysis platform (https://www.isanger.com/) provides specific steps for the analysis.The raw sequencing data were deposited in the NCBI Sequence Read Archive under project accession code SRP241743.
3 RESULT
3.1 Environmental parameters
Supplementary Table S1 presents the physicochemical characteristics of water and sediment samples collected in diff erent sites in the reservoir.There were some diff erences in the physicochemical indicators among the five water samples, but the environmental variables among the five sediments were not significantly diff erent. In addition,Supplementary Fig.S1 reveals the ecological purification eff ect on TN and TP in diff erent functional zones of the reservoir. Overall, the TN concentration from the inlet (JZW1) to the ecological sustaining zone (JZW5) was reduced by 47.6% and the TP concentration by 56.2% (Supplementary Fig.S1). In addition, the pH of all the studied samples was nearly neutral (7.59 to 7.65), the temperature was between 29.3 and 30.3 °C, and the concentrations of Chlaranged 8.65-11.94 mg/m3(Supplementary Table S1).Compared to those in water samples, the concentrations of TP and TN in sediments were higher. Furthermore,the concentrations of DO were higher in JZW4 than in JZW2, and the TN and TP concentrations in JZW5 were lower than those in JZW1 (Supplementary Table S1).
3.2 Analysis of bacterial community diversity
3.2.1 Alpha diversity analysis
From the five water and five sediment samples,410 047 raw sequences were produced by paired-end sequencing of the 16S rRNA gene. After quality filtering and removal of singletons and chimeras,112 994 eff ective sequences were obtained. These eff ective sequences were normalized for analysis of diversity indices. In all samples, the total number of OTUs observed was 3 483 (Supplementary Table S2).Library coverage for all the samples was also satisfactory (>99%), indicating that the OTUs of each bacterial library were well captured (Supplementary Fig.S2). In addition, the water samples had lower OTU richness than the sediment samples.
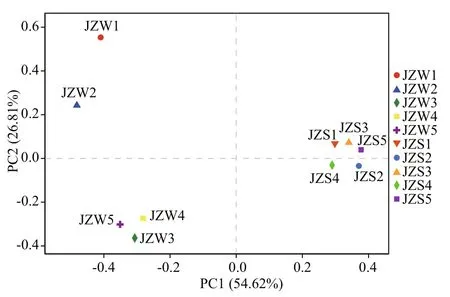
Fig.2 Principal coordinate analysis (PCoA) showing the diff erences in water (JZW1-JZW5) and sediment(JZS1-JZS5) bacterial community compositions in the Jinze Reservoir
The Shannon indices were 1.673-4.426 and 5.004-5.168 for the water and sediment samples, and the Chao 1 indices of the water and sediment samples were 324.4-472.5 and 396.7-427.5, respectively(Supplementary Table S2). The Shannon diversity indices and Chao1 index of the sediment samples showed little spatial fluctuations. Compared with water samples (Shannon average of 3.395), the diversity indices (Shannon average of 5.072) were higher in sediments. In addition, among all the samples, sample JZW4 showed the most diverse bacterial community (Chao1=472.5) (Supplementary Table S2).
3.2.2 Beta diversity analysis
The results of PCoA revealed heterogeneity in bacterial community between water and sediment in diff erent locations at the OTU level (Fig.2). Two PCoA axes accounted for 81.43% of the total variation in the bacterial community composition. When water and sediment samples were analyzed together, the bacterial community composition in the water samples from five sites were significantly diff erent from those of sediment samples, and those of the sediments were similar. In addition, samples JZW3,JZW4, and JZW5 were clustered in the same quadrant in high similarity (R=0.78;P<0.01).
3.3 Bacterial community composition
The taxonomic compositions of the water and sediment samples from the reservoir at phylum level are depicted in Fig.3a. For bacteria in water sample,Proteobacteria were the most abundant, with a relative abundance of more than 70%, of which Betaproteobacteria (up to 47.5%) was the main member of Proteobacteria, followed by Gammaproteobacteria (22.1%) (Fig.3b). The second major phylum was Cyanobacteria, taking 7.90% of the total. At all sampling sites, the cumulative abundance of the two most dominant phyla was greater than 80%. In addition, Bacteroidetes (6.85%),Actinobacteria (7.10%), Planctomycetes (2.15%),and Chloroflexi (1.68%) were the other four commonly detected phyla (Supplementary Fig.S3). In water samples, the relative abundance of bacterial communities between JZW2 and JZW4 was statistically significant (P<0.001) (Fig.4a). The relative abundance of Proteobacteria in JZW5 was significantly lower than that in JZW1 (Fig.3a).However, Actinobacteria were significantly more prevalent in JZW5 than in JZW1 (Fig.3a). In addition,the relative abundance of Cyanobacteria was significantly lower in JZW5 than in JZW4, but a higher relative abundance of Bacteroidetes was observed in JZW2.
The common phyla shared in all sediment samples included Proteobacteria (56.25%), Bacteroidetes(9.92%), Chloroflexi (8.24%), Acidobacteria (7.18%),Nitrospirae (5.53%), and Cyanobacteria (1.57%).Compared with the water bacterial communities, the relative abundance of sediment bacterial communities between JZS2 and JZS4 was not statistically significant (Fig.4b). Proteobacteria were significantly more in JZS1 (inlet) (60.97%) than in JZS5 (ecological sustaining zone) (51.76%) (Fig.3a), but the relative abundance of Nitrospirae in JZS5 was significantly lower than that in JZS4 (ecological purification zone)(Fig.3a). Moreover, Bacteroidetes dominated in JZS2(pretreatment zone), while Acidobacteria was significantly detected in JZS4. Betaproteobacteria and Gammaproteobacteria were the main classes of Proteobacteria in all sediment samples (Fig.3b).
Figure 5 shows the taxonomic composition of the water and sediment bacterial communities in the Jinze Reservoir at genus level. The taxa belonging toMethylotenerawere the most in abundance,accounting for 24.82% of the sequences, followed by unclassified Comamonadaceae (11.23%),Pseudomonas(8.65%),Acinetobacter(7.94%), and unclassified subsection (4.25%) in water samples.The dominant taxa included unclassified Xanthomonadalesincertaesedis(10.68%),unclassified Anaerolineaceae (6.51%),Nitrospira(5.53%), norank Acidobacteria (5.26%), and norank Alcaligenaceae (4.04%). The distribution of each dominant genus in the water and sediment samples was highly diversified among the five sampling sites.For the water bacterial communities, the genushgcIclade was dominant in JZW3, JZW4, and JZW5,while the generaMethylotenera(67.49%) andPseudomonas(24.11%) were dominant in JZW1(Fig.5). However, a higher abundance of the genusNitrospirawas observed in the sediment samples, but this genus was not detected in the water samples, and the relative abundance of the genusNitrospirawas lower in JZS5 than in JZS4 (Fig.5).
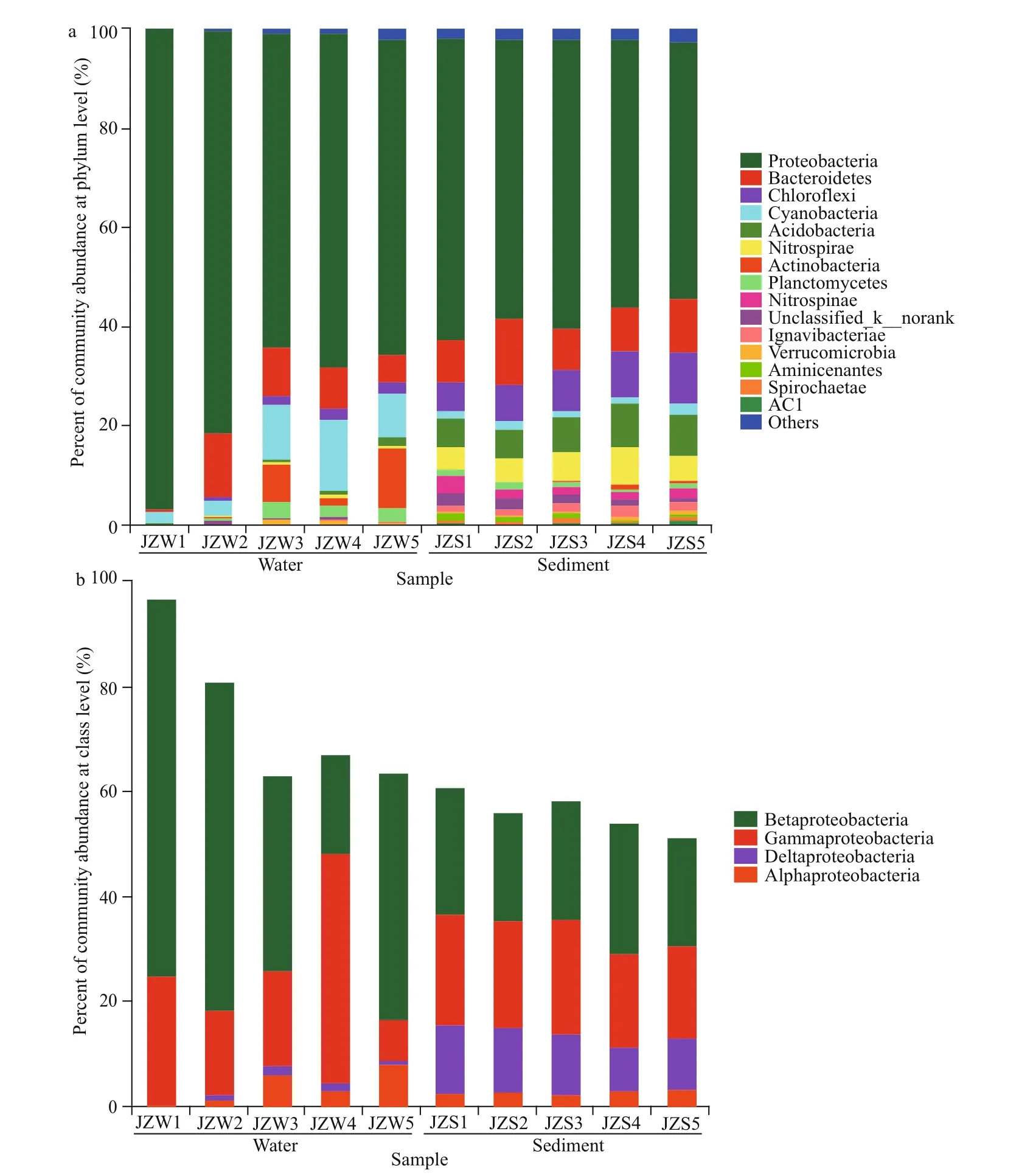
Fig.3 Comparisons of the bacteria taxonomic profiles of water and sediment samples from the Jinze Reservoir in relative abundance of the OTUs
3.4 Redundancy analysis
The correlation between physical and chemical indicators and the water and sediment bacterial communities of the Jinze Reservoir was determined by RDA. The positions of the five samples were relatively dispersed, indicating that they had diff erent bacterial community compositions. The physical and chemical properties of the water on the two RDA axes explained 86.27% and 8.69% of the total bacterial composition variation in water at phylum level(Fig.6a). TN was an influential factor that aff ected the distribution of bacteria in the water and contributed to the relationship between bacterial assemblage and environment. All Cyanobacteria, Chloroflexi, and Actinobacteria showed negative correlations with TN. However, Proteobacteria showed positive correlations with TP, but negative correlations with DO (Fig.6a). For sediment communities,environmental factors explained 91.57% of the sediment bacterial composition. Proteobacteria and Nitrospirae showed positive correlations with TP and TN (Fig.6b). Moreover, Acidobacteria was negatively correlated with TN (Fig.6b).
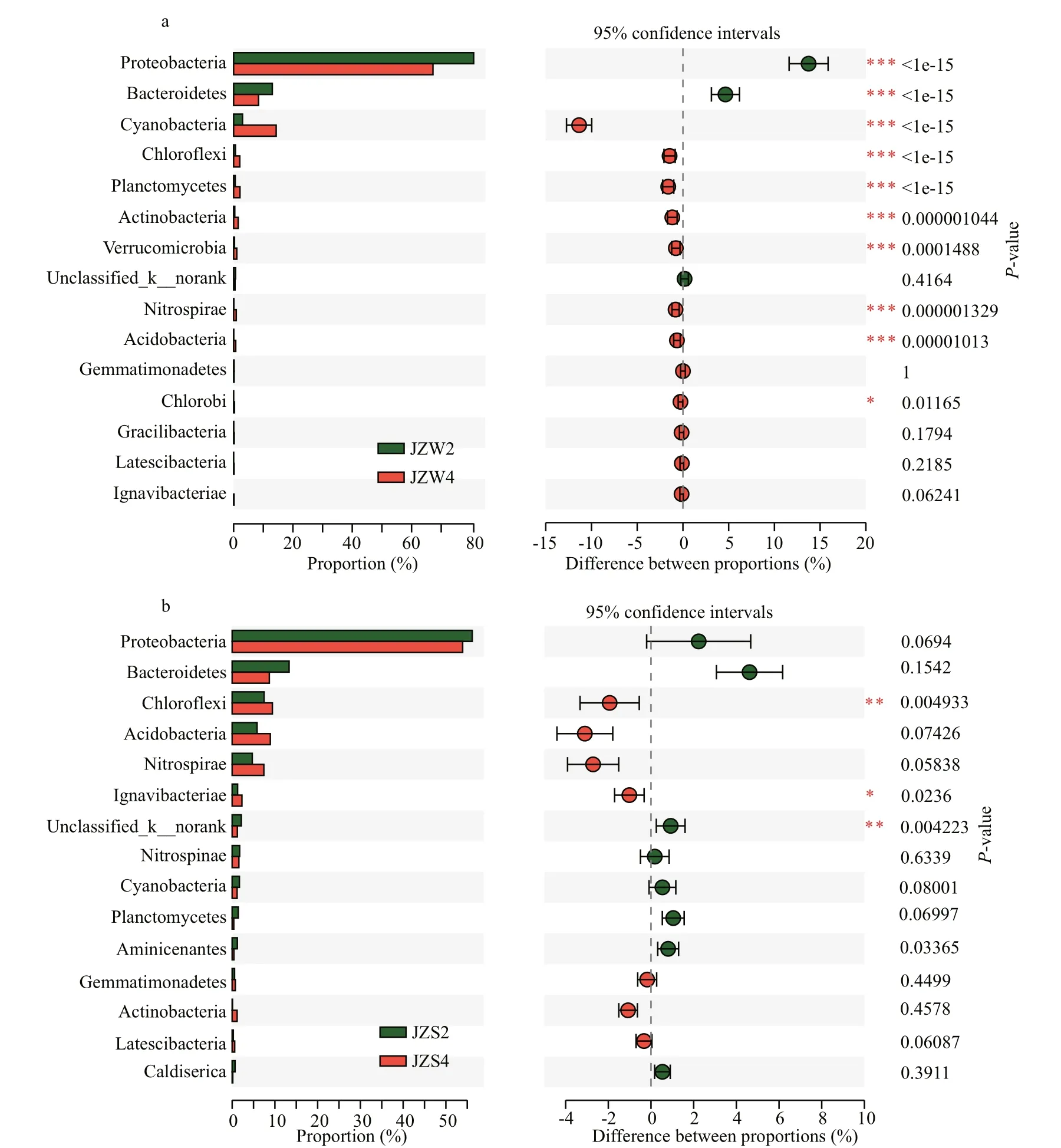
Fig.4 The diff erence in relative of taxa abundance between pretreatment zone and ecological purification zone
3.5 Co-occurrence network analysis
Co-occurrence networks of bacteria in water and sediment samples were built based on correlation relationships (Fig.7a & b). In the sediment bacterial co-occurrence network, 296 nodes were connected at 3 212 edges. The clustering coeffi cient was 0.65.Denser co-occurrence network was found in the water than in the sediment. The water bacterial network consisted of 274 nodes and 4 143 edges. The high clustering coeffi cient (0.77) also suggest that the bacterial network was closely connected.
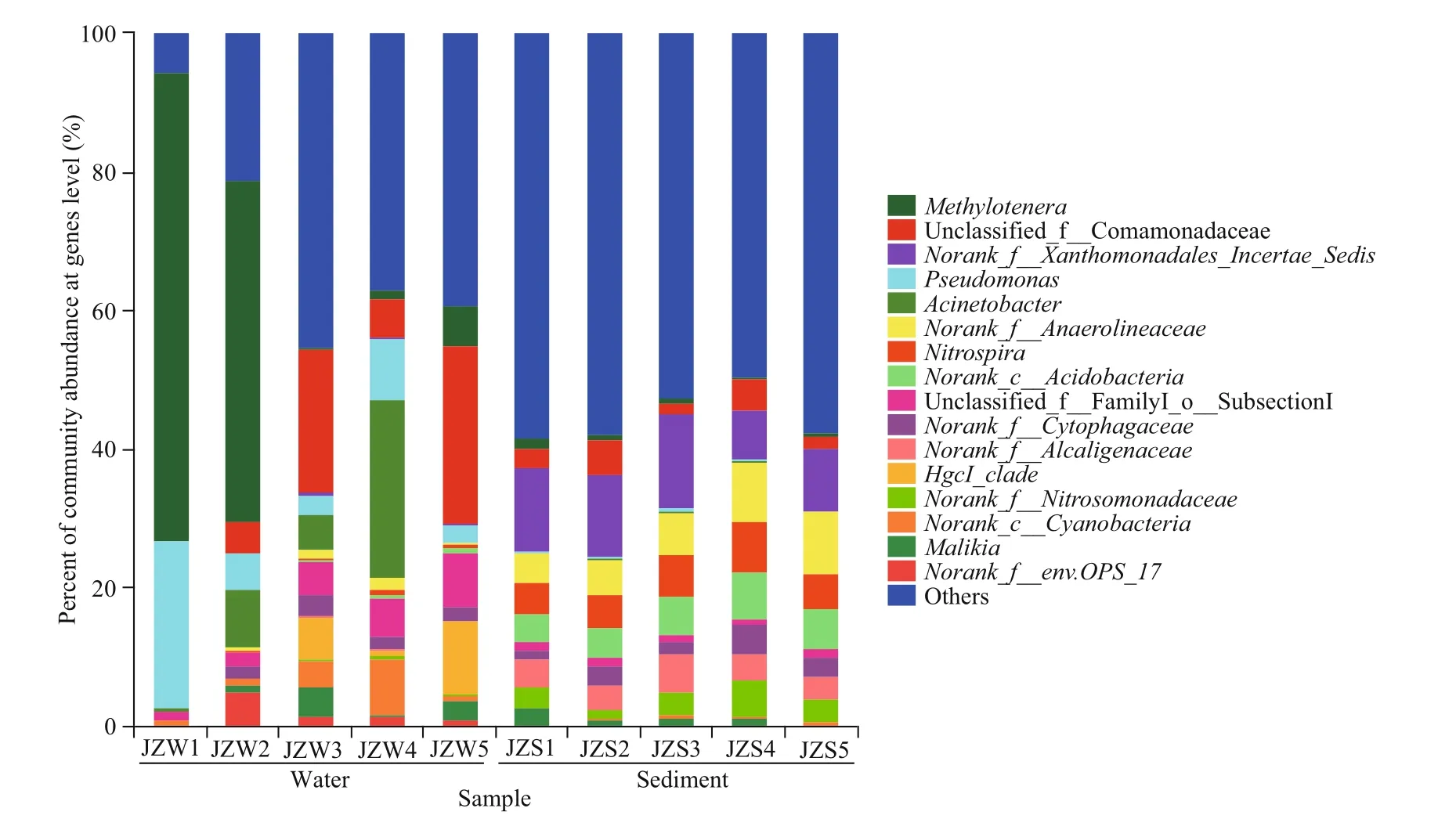
Fig.5 Bacteria community composition at the genus level in water and sediment samples
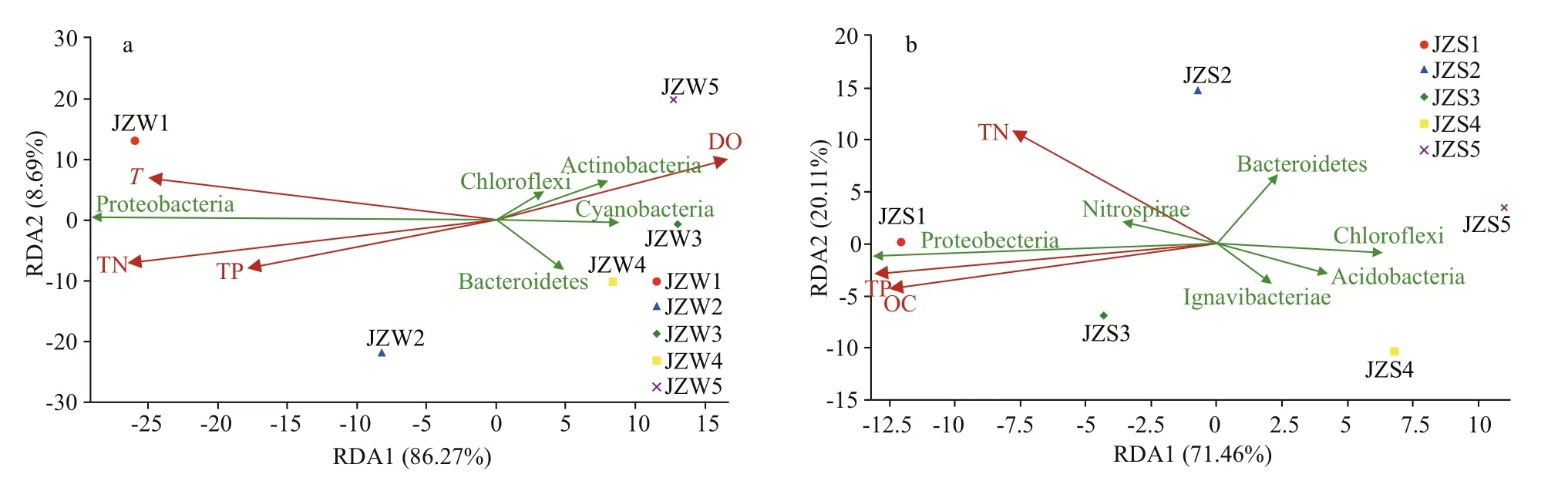
Fig.6 Redundancy analysis of bacterial community composition and physicochemical variables in water samples (a) and sediment samples (b)
All genera in the bacterial networks of waters and sediments were assigned to bacterial phyla. Among them, Proteobacteria and Bacteroidetes accounted for the two largest proportions in the water bacterial network (Fig.7a), and six phyla were widely distributed in the sediment bacterial network:Proteobacteria, Bacteroidetes, Actinobacteria,Chloroflexi, Planctomycetes, and Nitrospirae (Fig.7a).When the distribution of nodes was modularized, all the nodes of the water and sediment bacterial networks were divided into six main modules (>10 nodes)(Fig.7b). Conversely, taxonomic relatedness was clearly a key factor in determining the modular composition in the network. Typical examples were the genushgcIclade andCL500-29marine group,which dominated in module I of the water bacteria network, while module I of the sediment bacteria network was dominated by the generaNitrospiraandPseudomonas. In addition, according to the betweenness centrality scores, the key taxa in the identified water bacteria network werePseudomonasand unclassified Comamonadaceae, butIgnavibacteriumandVariovoraxwere identified as the key taxa in the sediment bacteria network. This result suggests that the bacteria were the major taxa in the co-occurrence network.
3.6 Bacterial metabolism
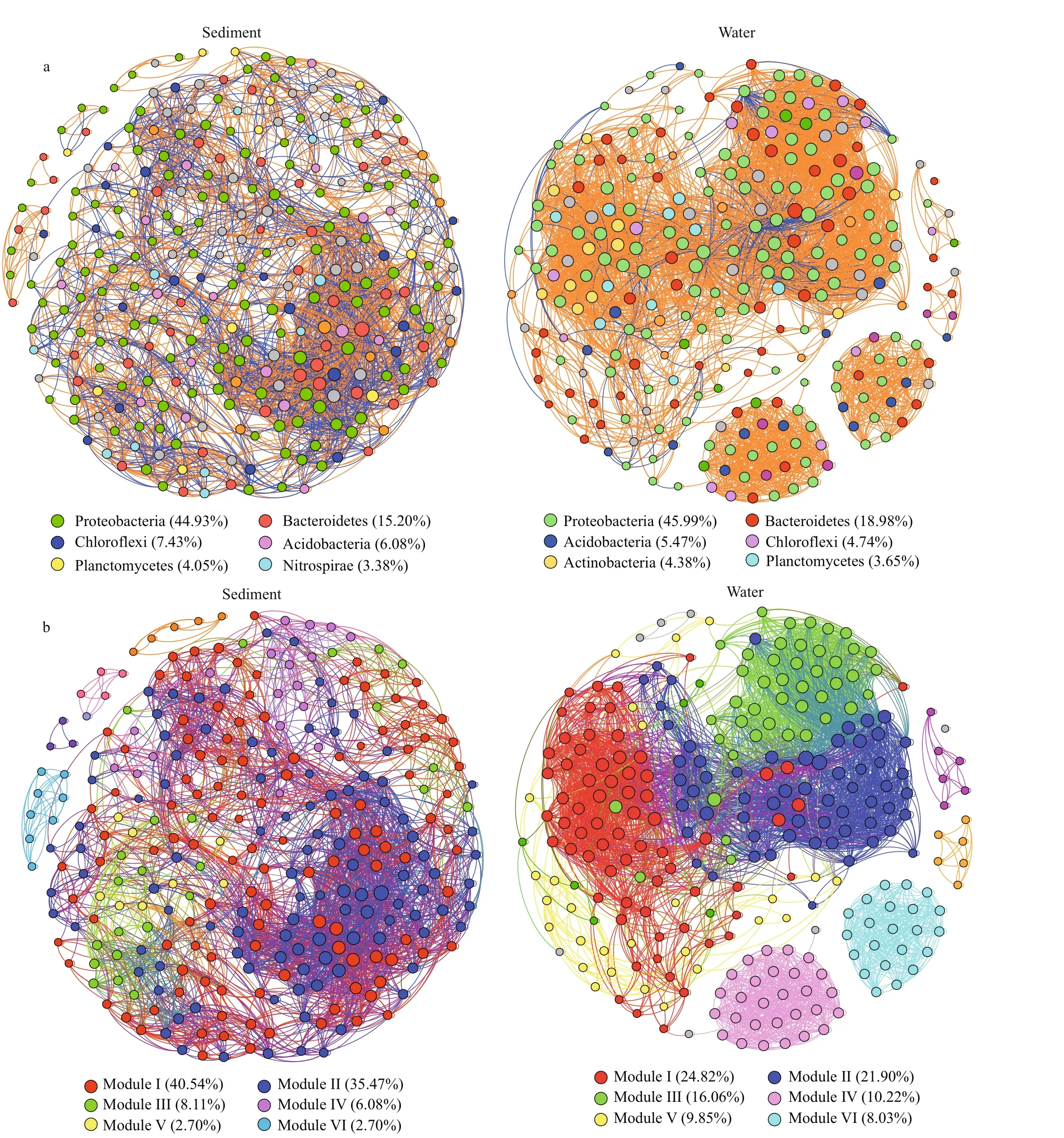
Fig.7 The co-occurrence networks
PICRUSt software was used to predict the functional contributions of bacteria in reservoir samples. The results show that bacterial communities potentially participated in a variety of basic processes and showed extensive genetic diversity (Figs.8 & 9).In KEGG level 2, metabolism was the most abundant functional classes (Figs.8 & 9). The most common predicted metabolic genes present in these bacterial communities participated in amino acid metabolism and carbohydrate metabolism. For sediment bacteria,the predicted genes abundance in carbohydrate metabolism in JZS4 was lower than in JZS1, and the predicted genes abundance in membrane transport was higher in JZS5 of the reservoir (Fig.9). In addition, a variety of biodegradation pathways of xenobiotic compounds was observed in all reservoir samples (Figs.8 & 9). For water bacteria, the predicted genes abundance of xenobiotic biodegradation decreased from that in JZW1 to that in JZW5 (Fig.8).In addition, as key agents in Polycyclic Aromatic Hydrocarbons (PAH) biodegradation among various predicted genes that involved in xenobiotic degradation in water samples, the predicted genes that related to benzoate biodegradation increased in JZW1 from those in JZW4 (Fig.8). In the sediment, however,the number of predicted genes affi liated with biodegradation pathways of xenobiotic compounds was relatively constant (Fig.9).
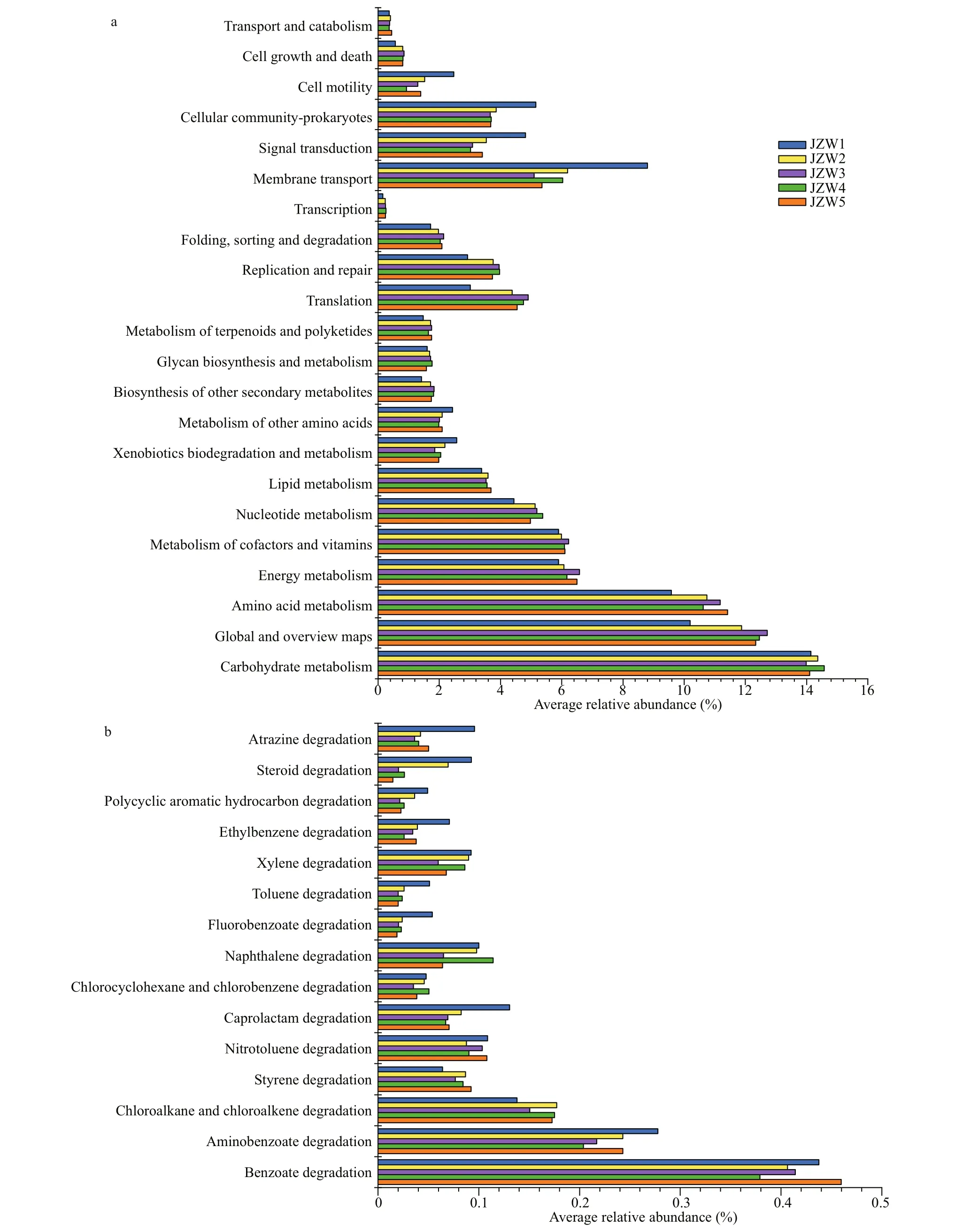
Fig.8 PICRUSt metabolic predictions (a) and xenobiotic biodegradation metabolic predictions (b) of the bacterial communities in water samples
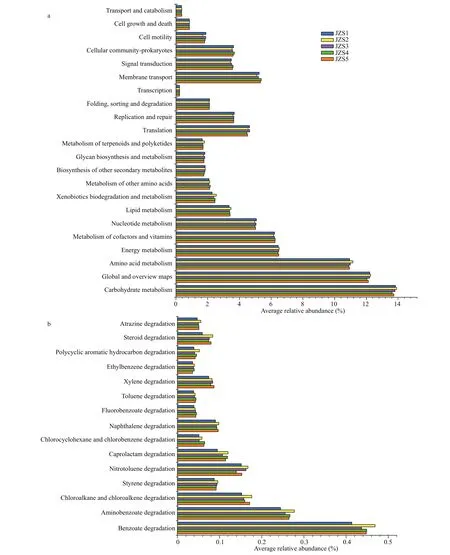
Fig.9 PICRUSt metabolic predictions (a) and xenobiotic biodegradation metabolic predictions (b) of the bacterial communities in sediment samples
4 DISCUSSION
Reservoirs as an important water source of artificial drinking water for cities, the maintenance and improvement of water quality are crucial. However,outbreaks of algal bloom in reservoirs are well documented worldwide (McLaughlan and Aldridge,2013). Relevant research has shown that biological regulation and ecological restoration technology are eff ective measures to control nutrient levels(McLaughlan and Aldridge, 2013). In this study, the TN concentration in the Jinze Reservoir after passing through the ecological purification zone was reduced by 16.7%, while the TP concentration was reduced by 33.3% (Supplementary Fig.S1). As the core unit of the water ecosystem restoration, an ecological purification zone makes use of plant purification and bacterial decomposition to remove pollution (Qian and Chen, 2016). As well, the ecological purification zone in the Jinze Reservoir showed purification eff ect on TN and TP removal from the water.
The reservoir presented a typical freshwater bacterial community. The core bacterial communities in water and sediment were composed of mainly Proteobacteria, Bacteroidetes, Chloroflexi,Cyanobacteria, and Acidobacteria (Fig.3a). These indigenous phyla in the reservoir were consistent with those of other studies (Bai et al., 2014; Ibekwe et al.,2016). Moreover, to study the heterogeneity among the bacterial communities, PCoA was used and revealed a significant compositional diff erence in bacterial community in water and sediment (P<0.01)(Fig.2). The abundance of Acidobacteria in the sediment samples was significantly higher than that in the water samples (Fig.3a), which is a normal case as shown in a previous study that Acidobacteria are ubiquitous in freshwater sediments and are the main members of bacterial communities (Jones et al.,2009). Proteobacteria represented the most abundant phylum in water and sediments of the Jinze Reservoir.The high abundance of Proteobacteria indicated that they played a key role in the biogeochemical functioning and processes of reservoir water and sediment (Chen et al., 2017; Mao et al., 2019). Further analysis revealed that the dominant class in phylum Proteobacteria diff ered among samples (Fig.3a).Betaproteobacteria dominated in the water of the reservoir, being typical, dominant, and crucial freshwater bacterial assemblages in aquatic habitats,which is similar to those of previous studies (Brümmer et al., 2003; Crump and Hobbie, 2005; Salcher et al.,2013), whereas Gammaproteobacteria and Betaproteobacteria dominated in the sediment samples (Fig.3b), which supports previous findings that Betaproteobacteria are able to degrade complex organic macromolecules in sediments (Kirchman,2002) and Gammaproteobacteria are present in organic-rich sediments (Dang and Lovell, 2016).
In this study, significant diff erences were observed in the Proteobacteria and Actinobacteria of the reservoir between JZW2 (pretreatment zone) and JZW4 (ecological purification zone) (P<0.001)(Fig.4a). Some studies found that the cultivation of aquatic plants has a positive impact on water bacterial community diversity (Shehzadi et al., 2014; Lv et al.,2017), which indicates that this strategy aff ects the water community composition by increasing the bacteria diversity in water. Furthermore, the relative abundance of Proteobacteria obviously decreased from JZW1 (inlet) to JZW5 (ecological sustaining zone), and the pronounced increase of Proteobacteria was positively correlated with the TP, which is consistent with previous research results that Proteobacteria are aff ected mainly by TP content, and high abundance of Proteobacteria depends on high nutrient availability (Fierer et al., 2007; Zoppini et al.,2010). In contrast, higher relative abundance of Actinobacteria in the reservoir water was determined in JZW5 (Fig.3a), and the TN concentration in JZW5 was low (Supplementary Table S1), which is due to the decrease in relative abundance of Actinobacteria with the increase in nutrient concentration (Benveniste and Davies, 1973; Goodfellow and Williams, 1983).
To fully understand the bacterial processes and material cycling in reservoir ecosystems, it is necessary to study the community composition of sedimentary bacteria (Keshri et al., 2018). The abundance of Bacteroidetes was higher in overall(Fig.3a), but relatively lower in JZS4 than in JZS2.Bacteroidetes are gram-negative bacteria that are mostly heterotrophic; they can further settle and decompose complex organic matter (Tang et al.,2017). In this study, it is not surprising that Bacteroidetes were widespread in organic-rich JZS2.Nitrospiracontains nitrite-oxidizing bacteria as a part of the biogeochemical nitrogen cycling. The RDA results show thatNitrospirawas positively correlated with TN (Fig.6b), which fully supported thatNitrospirais strong indicator to nitrogen cycling(Huang et al., 2017). However, Acidobacteria were negatively correlated with TN (Fig.6b), supporting previous reports that the distribution of Acidobacteria is negatively correlated with low nutrient availability.
Network analysis can provide deep and unique insights into highly complex microbial communities in an ecosystem (Ren et al., 2019). Exploring cooccurrence patterns to understand bacterial interactions (Jiao et al., 2016) and nonrandom cooccurrence in ecological purification reservoirs can illustrate the role of deterministic processes in the community composition. Analysis of the bacterial networks in the water and sediment samples showed that the most nodes belonged to Proteobacteria,Bacteroidetes, and Actinobacteria (Fig.7a & b). The nodes of the network of both water and sediment bacteria could be divided into six main modules.Nodes in diff erent modules performed with diff erent functions (Newman, 2006) and closely-related taxa tended to be jointed and clustered, which reflects strong ecological links among species within the taxa(Chaff ron et al., 2010; Ju and Zhang, 2015), which was obvious in the network module. For example,Module I in the water bacterial network was mainly assembled by OTUs that affi liated with Actinomycetales (hgcIclade andCL500-29marine group) that are abundant in nutrient-poor niches(Hahn et al., 2003; Warnecke et al., 2005; Allgaier and Grossart, 2006), and rich bacteria of Module I in the water bacterial network were found in low nutrient concentrations (as shown in JZW5). Furthermore, a higher relative abundance of the genushgcIclade was observed in JZW5 (Fig.5), suggesting that the genushgcIclade in JZW5 with low nutrient concentrations was more competitive than the other bacteria presented. However, rich bacteria in Module I in the sediment bacterial network were mainly found in JZS1 (inlet) and JZS2 (pretreatment zone). The main taxa in Module I of the sediment bacterial network might be bacteria involved in biogeochemical nitrogen cycles. For example, the genusNitrospiracontained nitrite-oxidizing bacteria as a part of the biogeochemical nitrogen cycles (Altmann et al.,2003). GenusPseudomonashas been identified as a main functional bacterial phylotype in nitrogen cycles(Toukdarian and Lidstrom, 1984). These results indicate that Module I in the sediment bacterial network was related to nitrogen cycling.
Identifying the modules that maintain network connectivity with betweenness centrality highlights its application in defining the key species of the system (Vick-Majors et al., 2014; Banerjee et al.,2016a). The higher betweenness centrality scores of the generaPseudomonasand unclassified Comamonadaceae observed in the water bacterial network indicated that these genera might play key roles in maintaining ecological community composition and functions. The generaPseudomonasand unclassified Comamonadaceae were the main taxa in the water samples. Some members of thePseudomonasgenus have previously been shown to metabolize chemical pollutants in the environment(Yen et al., 1991; O’Mahony et al., 2006). These features resulted in a high dominance of the genusPseudomonasin JZW1 (inlet) with high nutrient concentrations. However, the genus unclassified Comamonadaceae showed a lower abundance in the nutritionally deficient JZW4 (ecological purification zone) (Fig.5), potentially because members of the genus unclassified Comamonadaceae often become dominant groups in nutrient-rich environments.Additionally, the generaVariovorax,Dechloromonas,and unclassified Comamonadaceae produced higher betweenness centrality scores in the sediment bacterial network in this study. However, we noted that the abundance of these genera was less than 1% (Fig.5).At the genus level, the genusVariovoraxwas identified as the prominent phylotype. The prominence of this genus might be due to the high rates of substrate uptake and high mortality rates of bacterivory in unfavorable habitats (Abou-Shanab et al., 2007;Satsuma, 2010; Kasalický et al., 2013). Moreover, the genusDechloromonashas generally been recognized as having high effi ciency in phosphorus removal(Zhou et al., 2012), supporting the importance of a rare genera in bacterial communities.
PICRUSt was used to assess the potential function of bacterial communities in the reservoir, and to predict the occurrence of potentially participated in fundamental processes such as cellular processes and metabolism in bacterial communities (Figs.8 & 9).The predicted genes related to carbohydrate metabolism were higher in sediment bacteria in JZS1 at the inlet (Fig.9) with high nutrient concentrations(Supplementary Table S1), which reflected the physicochemical properties of the river water in which the metabolic activities of sediment bacteria were promoted (Lu et al., 2017). The metabolism of sediment bacteria increased to adapt to adverse environmental conditions in JZS1 (inlet) with a high nutrient concentration. In addition, the predicted genes related to membrane transport were higher in sediment bacteria at a lower nutrient concentration in JZS5 (ecological sustaining zone) of the reservoir(Fig.9), accompanied by a relatively lower abundance of genus unclassified Comamonadaceae, which is common in nutrient-rich environments. These findings support previous reports showing that genes related to membrane transport are involved in environmental information processing and nutrient acquisition, and they can be generated in suffi cient amounts only under nutritionally restricted conditions(Williams and Cavicchioli, 2014). However, the predicted gene abundance of xenobiotic biodegradation in the water samples was relatively lower in JZW5 (Fig.8). These results show clearly the presence of a certain correlation between the genes abundance for xenobiotic degradation and xenobiotic biodegradation rates (Pagé et al., 2015; Kansole and Lin, 2016). The predicted abundance of most genes related to xenobiotic biodegradation showed a decreasing trend across reservoir from inlet to outlet as shown in the water samples, which might be due to the higher concentration at the inlet. In addition, the predicted gene abundance of benzoate biodegradation was lower in JZW4 than in JZW1 (Fig.8), which was also accompanied by the decrease in the abundance of potential PAHs-degrading bacteria; for example,genusPseudomonasin water bacteria showed a lower abundance in JZW4 (Fig.5). Available studies indicate that the enrichment ofPseudomonasmay suggest the accumulation of environmental pollutants (Yen et al.,1991; O’Mahony et al., 2006). Therefore, these features led to a decrease in the predicted genes abundance of benzoate biodegradation in water bacteria in JZW4 in downstream of the ecological purification zone.
5 CONCLUSION
In this study, we revealed changes in environmental parameters and the spatial changes in bacterial communities in waters and sediments in diff erent functional zones of the Jinze Reservoir, an ecological purification reservoir. The results present that the concentrations of TN and TP in the reservoir decreased obviously from the inlet to the outlet as shown in the water samples. Although both water and sediment bacterial communities shared similar bacterial communities, diff erences in the bacterial communities between the water and sediment habitats were obvious. Nonrandom co-occurrence and modular patterns were observed in the bacterial communities,providing a new perspective regarding bacterial assembly in ecological purification reservoirs. In addition, diff erences were observed in the abundance of predicted genes associated with metabolism in bacteria throughout in the reservoir. These results suggest that studying bacterial communities using water and sediments in diff erent functional zones in ecological purification reservoir is essential to obtain a comprehensive bacterial community profile to well manage water quality control.
6 DATA AVAILABILITY STATEMENT
All sequence data were deposited in the National Center for Biotechnology Information (NCBI)Sequence Read Archive with accession No.SRP241743.
 Journal of Oceanology and Limnology2021年4期
Journal of Oceanology and Limnology2021年4期
- Journal of Oceanology and Limnology的其它文章
- Numerical study of the seasonal salinity budget of the upper ocean in the Bay of Bengal in 2014*
- Study on evaluation standard of uncertainty of design wave height calculation model*
- A fast, edge-preserving, distance-regularized model with bilateral filtering for oil spill segmentation of SAR images*
- A Gaussian process regression-based sea surface temperature interpolation algorithm*
- Climatology and seasonal variability of satellite-derived chlorophyll a around the Shandong Peninsula*
- Sources of sediment in tidal flats off Zhejiang coast, southeast China*