
三七不同来源指纹图谱数字化研究
2021-07-29 04:59童湘伟石坤连梁业婷易延逵
医学信息 2021年14期
童湘伟,石坤连,梁业婷,沈 颖,周 蓓,易延逵
(1.涟源市人民医院药剂科,湖南 涟源 417100;2.南方医科大学中医药学院,广东 广州 510515;3.广西中医药大学药学院,广西 南宁 530001)
三七为五加科多年生草本植物三七[Panax notoginseng(Burk.)F.H.Chen]的干燥根和根茎,具有散瘀止血,消肿定痛的功效,用于各种原因导致的出血,淤血和跌打损伤[1]。三七的主要活性成分是三七总皂苷(panax notoginseng saponins,PNS)[2],研究主要以该类成分为参照物建立指纹图谱。据相关文献记载[3,4],已有学者对三七总皂苷的提取、分离纯化的方法和技术进行了研究。这些成果为其指纹图谱和药理的研究奠定了基础。中药指纹图谱[5]是指将中药适当处理后,应用特定分析手段获取能够表示其特性的图谱,截止2020 年5 月,大批学者对三七指纹图谱的建立做了研究,包括对三七HPLC 指纹图谱进行了较多研究[6-8]、采用HPLC 指纹图谱来鉴别人参、西洋参及三七[9]、利用高效液相联用质谱技术建立三七药材的指纹图谱[10]以及借助主要成分分析和聚类分析方法比较不同批次、产地的三七药材质量的差异性,为全面评估三七药材质量提供参考[11]。本文在前期对指纹图谱研究基础上,针对天然产物成分的复杂性,提出了包括“全指纹图谱系”“区间指纹图谱”的构建等,同时考虑针对目前指纹图谱研究存在的问题,提出了进行中药指纹图谱峰值的数字化问题研究[12]。本次主要对三七指纹图谱和考察不同来源三七指纹图谱的波动范围进行探索。
1 仪器与材料
1.1 仪器设备 岛津高效液相色谱仪(岛津<苏州>有限公司),SK1200H 型超声器(上海科导超声仪器有限公司),DFY-300 摇摆式多功能高速中药粉碎机(温州顶历医疗器械有限公司),MSX(SD EE)型电子天平(Sartorius,Goetiingen,Germanny),BL-600 型电子天平(厦门佰伦斯电子科技有限公司)。
1.2 材料及药品 对照品:三七皂苷R1(中国药品生物制品检定所,批号110745-200415,供含量测定用)、人参皂苷Re(中国药品生物制品检定所,批号110754-200320,供含量测定用)、人参皂苷Rg1(中国药品生物制品检定所,批号22427-39-0,供含量测定用)。药材:三七(批号:S9219506,购自广东省药材公司中药饮片厂)、三七(批号:S9218412,购自广东省药材公司中药饮片厂)、三七(批号:190705,购自广东省采芝林医药公司)、三七(批号:180907,购自广东省采芝林医药公司)、三七(批号:180608,购自四川薪菊花中药饮片股份有限公司),上述三七药材经南方医科大学中医药学院的刘传明教授鉴定均为五加科三七的干燥根和根茎。试剂:D-101 型大孔吸附树脂(批号:HC191026,和诚新材料有限公司);甲醇、乙腈为色谱纯,水为重蒸水,其它试剂均为分析纯。
2 方法与结果
2.1 色谱条件 色谱柱[9,13,14]:InertSustainTM AQ-C18(250 mm×4.6 mm,5 μm),流动相:乙腈-0.1%磷酸水溶液,梯度洗脱见表1,流速:1.0 ml/min,检测波长:203 nm,柱温:25 ℃,进样量10 μl,记录时间为70 min。

表1 梯度洗脱程序
2.2 供试品溶液的制备 精密称取三七药材粉末(过60 目筛)2.0 g,置于锥形瓶中,加入70%乙醇50 ml,浸泡30 min,80 ℃恒温水浴加热冷凝回流提取1.5 h,过滤,滤液浓缩至2 ml 左右,与适量大孔树脂(95%乙醇浸泡至无气泡)混合、拌匀,挥干。D-101 型大孔树脂柱色谱分离纯化,用95%乙醇大孔树脂浸泡3~4 h 至无气泡,在色谱柱底部垫一小团棉花,装大孔树脂层10 cm 左右,纯净水洗至无醇味,压平大孔树脂柱层面,加入样品,放一团棉花。洗脱,先用200 ml纯净水洗脱,水洗脱液弃去,再用60 ml 80%乙醇洗脱[3],洗脱流速为60 滴/min,乙醇洗脱液浓缩至干,用70%甲醇溶解,用5 ml 容量瓶定容,混合均匀,通过0.45 μm 的微孔滤膜,即得。
2.3 参照物的选择和制备 对照品溶液的制备:分别精密称取三七皂苷R1、人参皂苷Re、人参皂苷Rg1等三种对照品,用70%的甲醇配制成每5 ml 均含0.4 mg 的一份混合对照品溶液和三份单一对照品溶液。
2.4 测定法 分别精密吸取参照物溶液与供试品溶液各10 μl,注入液相色谱仪,测定,记录70 min 的色谱图,即得。以分离度好的、峰形适宜的三七皂苷R1的图谱峰即8 号峰为参照物峰,记为S 峰,见图1。
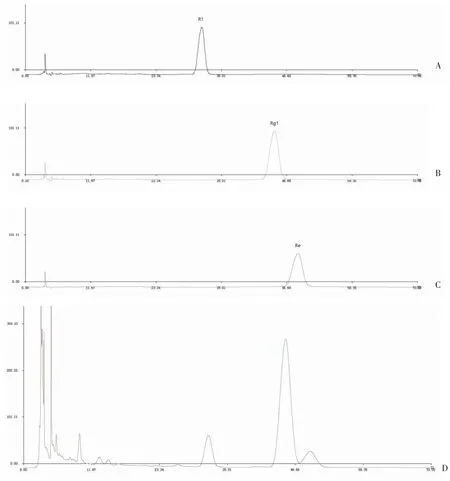
图1 参照物色谱图与三七药材共有指纹峰指纹图
2.5 三七指纹图谱的建立 取10 批次同一来源药材粉末各1 份,按“2.2”项下的方法制备供试品,“2.1”项下的色谱条件测定指纹图谱,采用“中药色谱指纹图谱图相似度评价系统软件(2004A 版)”进行处理,以全谱和峰面积进行匹配,以第六批即S6 色谱图为参照,完成自动匹配数据,生成对照图谱R,并以其为参照,完成相似度评价,10 批样品的相似度均在0.80 以上,见图2。共有指纹峰的标定:采用相对保留时间标定共有指纹峰,以图谱中三七皂苷R1为参照物,将各色谱峰保留时间与同一图谱中参照物的保留时间比较,其比值为各色谱峰的相对保留时间,计算10 批次指纹图谱中各色谱峰的相对保留时间。其中10 个色谱峰为各样品所共有,故标定它们为共有指纹峰。各共有指纹峰的相对保留时间分别为峰-1(0.1722~0.1769)、峰-2(0.2498~0.2535)、峰-3(0.2995~0.3041)、峰-4(0.4055~0.4109)、峰-5(0.4551~0.4604)、峰-6(0.5103~0.5141)、峰-7(0.8324~0.8346)、峰-S(1.0000)、峰-9(1.4172~1.4193)、峰-10(1.5492~1.5546)。同时以各将各色谱峰峰面积与同一图谱中参照物的峰面积比较,其比值为各色谱峰的相对峰面积,计算10 批次指纹图谱中各色谱峰的相对峰面积。共有指纹峰的相对峰面积分别为峰-1(0.1837~0.2777)、峰-2(0.0152~0.1104)、峰-3(0.3015~0.5118)、峰-4(0.1128~0.2807)、峰-5(0.0721~0.1343)、峰-6(0.0428~0.0499)、峰-7(0.0288~0.0638)、峰-S(1.0000)、峰-9(5.6985~5.8728)、峰-10(0.7872~0.8160)。
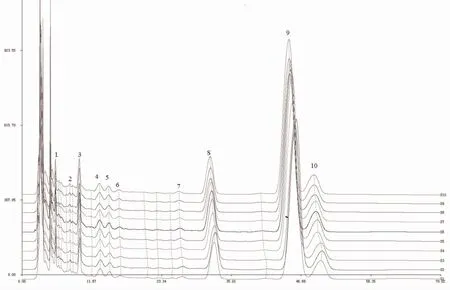
图2 相似度计算(全谱)图
2.6 方法学考查
2.6.1 精密度试验 取同一份供试品溶液,按“2.1”项下的色谱方法进行测定,连续进样6 次,计算各次指纹峰的相对峰面积和保留时间,结果RSD<3%,表明色谱仪器精密度良好,符合指纹图谱测定要求。
2.6.2 重复性试验 取同一批三七药材粉末6 份,按“2.2”项下的方法制备供试品溶液,按“2.1”项下的色谱方法进行测定,进样,计算各次指纹峰的相对峰面积和保留时间,结果RSD<2.5%,表明本试验重复性良好。
2.6.3 稳定性试验 取同一供试品,按“2.1”项下的色谱方法进行测定,分别在0、2、4、8、12、24 h 进样,计算各时间点的指纹峰的相对峰面积和保留时间,结果RSD<2.2%,表明供试品溶液在24h 内稳定性良好。
3 不同来源三七药材指纹图谱波动范围的考察
3.1 不同来源三七药材指纹图谱制定 取5 批不同来源的药材粉末各1 份,按“2.1”项下的色谱条件,“2.2”项下的方法制备供试品,制定指纹图谱。以图谱中三七皂苷R1 为参照物峰,将各色谱峰保留时间和峰面积与同一图谱中参照物的保留时间和峰面积比较,其比值为各色谱峰的相对保留时间,相对峰面积,图谱见图3。

图3 5 批不同来源的三七指纹图谱的比较图
3.2 不同来源三七药材指纹图谱波动范围 对共有峰的相对保留时间、相对峰面积归纳分析其波动范围。以各不同来源三七指纹谱各色谱峰保留时间和峰面积与同一图谱中参照物的保留时间和峰面积比较,其比值为各色谱峰的相对保留时间和相对峰面积。结果各共有指纹峰的相对保留时间分别为峰-1(0.1736~0.1784)、峰-2(0.2425~0.2548)、峰-3(0.2992~0.3056)、峰-4(0.4058~0.4134)、峰-5(0.4527~0.4597)、峰-6(0.5103~0.5151)、峰-7(0.8324~0.8344)、峰-S(1.0000)、峰-9(1.4167~1.4198)、峰-10(1.5497~1.5546)。各共有指纹峰的相对峰面积分别为峰-1(0.1567~0.2272)、峰-2(0.0922~0.2097)、峰-3(0.2402~0.4975)、峰-4(0.1569~0.1997)、峰-5(0.1082~0.1405)、峰-6(0.0487~0.0795)、峰-7(0.0494~0.0847)、峰-S(1.0000)、峰-9(4.0436~5.8524)、峰-10(0.4935~0.8973)。
4 讨论
在《中药注射剂指纹图谱研究的技术要求(暂行)》的通知中,对于中药注射剂原料来源上要求动、植物药材均应固定品种、药用部位、产地、采收期、产地加工和炮制方法,矿物药应固定产地和炮制、加工方法[13]。这就要求制定产品药材的指纹图谱要求来源加工工艺等要固定,对于不同来源加工工艺药材而言,要分别制定指纹图谱。本试验主要考察不同来源药材的波动范围,为其质量控制提供试验依据。本试验通过10 批次同一来源三七药材指纹图谱研究分析,制定了其指纹图谱;本试验通过5 批来源不同的三七考察指纹峰的波动范围;通过初步考察三七药材指纹峰波动范围,可以初步确定三七药材指纹图谱指纹峰波动范围如下:相对保留时间分别为峰-1(0.1730~0.1790)、峰-2(0.2400~0.2550)、峰-3(0.2990~0.3060)、峰-4(0.4050~0.4140)、峰-5(0.4520~0.4600)、峰-6(0.5100~0.5160)、峰-7(0.8320~0.8350)、峰-S(1.0000)、峰-9(1.4160~1.4200)、峰-10(1.5490~1.5550)。各共有指纹峰的相对峰面积分别为峰-1(0.1560~0.2280)、峰-2(0.0920~0.2100)、峰-3(0.2400~0.4980)、峰-4(0.1560~0.2000)、峰-5(0.1080~0.1410)、峰-6(0.0480~0.0800)、峰-7(0.0490~0.0850)、峰-S(1.0000)、峰-9(4.0430~5.8530)、峰-10(0.4930~0.8980)。
本结果共有峰的相对保留时间波动范围较小,而相对峰面积波动范围较大。共有峰的相对保留时间波动范围主要跟仪器方法等有关,因而其波动范围不会很大;相对峰面积波动范围跟成分的含量相关,不同来源的药材含量差异越大,波动范围就越大。因此,建立三七药材HPLC 指纹图谱的数字化波动范围对如何有效把控三七药材质量有一定的参考意义。本试验为药材的质量控制研究提供了可参考试验依据。但本试验试验样本数较少,试验使用的三七均产自云南,因而可进一步增加试验样本数,收集试验使用的三七药材产地尽量覆盖到三七主要产地。
以往建立三七指纹图谱的研究中,很少对三七提取液经过分离纯化后再建立指纹图谱。本试验参考了三七有效成分分离纯化的方法,选用D-101 型大孔树脂对三七药材提取液进行分离纯化[15]。将纯化前后的供试品得到的HPLC 指纹图谱进行比较,结果表明,纯化后建立的指纹图谱,5 min 前的峰高、峰面积会明显下降,5 min 后的峰的峰高和峰面积降低不明显,说明纯化有利于减少杂质的含量。
综上,本次初步探究获得的三七药材指纹图谱波动范围可对三七药材的质量控制研究提供试验依据。
猜你喜欢
数学小灵通(1-2年级)(2021年11期)2021-12-02
小哥白尼(趣味科学)(2021年11期)2021-02-28
小天使·一年级语数英综合(2020年10期)2020-12-16
中成药(2018年9期)2018-10-09
中成药(2017年9期)2017-12-19
中成药(2017年6期)2017-06-13
自动化学报(2016年8期)2016-04-16
青少年科技博览(中学版)(2015年7期)2015-08-12
中国药业(2014年16期)2014-05-14
恋爱婚姻家庭·青春(2009年9期)2009-10-26
