
西藏大花红景天EST-SSR开发及通用性分析
2021-07-26 10:44张力鹏滕彦娇王宏鹏李天宇宋文芹陈成彬
江苏农业科学 2021年11期
关键词:转录组
张力鹏 滕彦娇 王宏鹏 李天宇 宋文芹 陈成彬
摘要:以西藏大花红景天为研究对象,用MISA软件筛选转录组测序获得的48 790条unigene,开发EST-SSR(表达序列标签-微卫星序列)并对其通用性进行分析,以期为红景天属物种的遗传多样性研究和分子标记辅助育种奠定基础。结果表明,共检测出10 761个SSR位点,分布于8 973条unigene中,分布频率为22.06%,平均分布距离为4.725 kb。优势重复基序以单核苷酸、二核苷酸、三核苷酸为主,其中单核苷酸重复单元以基序A/T为主,共7 167个,二核苷酸重复单元以AG/CT为主,共1 008个。随机合成280对引物,其中248对表现出有效扩增,54对具有多态性。此外,SSR引物在长鞭红景天、狭叶红景天、菊叶红景天、圣地红景天、柴胡红景天、高山红景天中具有较高的通用性,扩增效率分别为75.0%、73.75%、72.5%、62.5%、70.0%、72.5%。
关键词:大花红景天;转录组;SSR;通用性分析
中图分类号: S567.23+9.01 文献标志码: A
文章编号:1002-1302(2021)11-0040-08
收稿日期:2020-08-29
作者简介:张力鹏(1989—),男,河北沧州人,博士,助理研究员,主要从事中药材种质资源品种改良研究。E-mail:nknanhai@163.com。
通信作者:陈成彬,博士,副教授,主要从事中药材种质资源与品种改良研究。E-mail:chencb@nankai.edu.cn。
红景天是双子叶蔷薇目(Rosales)景天科(Crassulaceae)红景天属(Rhodiola L.)植物,因其根或根茎的浸出液为红色而得名[1]。全世界红景天属植物共有90多种,其中我国占有主要的种质资源有70多种,主要分布在海拔(3 500~5 000 m)较高的山区,包括青藏高原及其毗邻带、横断山脉、天山山脉等[2-4]。其中以西藏占有的种类最多,为32种;四川次之,为22种;新疆为14种。红景天常被称作“高原人参”或“雪山仙草”应用于传统中医和藏医种,已经有数千年的历史。目前主要药用种为蔷薇红景天(R. rosea)、大花红景天(R. crenulata)、菊叶红景天(R. sachalinensis)、圣地红景天(R. sacra)、齿叶红景天(R. serrata)、高山红景天(R. cretinii)和长鞭红景天(R. fastigiata)等[5-7]。现代药理学研究发现,红景天根部含有40多种活性物质如红景天苷、没食子酸、苷原酪醇及其衍生物等,具有抗氧化、抗缺氧、抗辐射、抗衰老、抗疲劳、提高机体免疫力等作用。其中以大花红景天的活性物质含量最高、药效最好、使用最为广泛。因此《中华人民共和国药典》2015版中唯一指定的入药种为大花红景天[8]。
基于多聚酶链式反应(PCR)的分子标记技术具有多态性好、遗传性高、稳定性强、简单迅速的特点,已逐渐成为研究植物物种遗传多样性的有力工具[9]。其中简单重复序列(simple sequence repeat,简称SSR)也称微卫星,能够均匀、随机地分布于植物基因组DNA中,具有数量丰富、稳定性高和简单便捷等优点[10-12]。2013年,You等筛选分离了柴胡红景天的17对多态性引物以扩增微卫星位点(CCG)6、(AAG)8、(AGG)6、(CT)13、(AGC)6、(AC)10和(ATC)6,并将其成功应用于大花红景天、长鞭红景天和圣地红景天[13]。雷淑芸等基于高通量测序技术对唐古特红景天全基因组的SSR位点进行了分析,为红景天属植物SSR标记的开发奠定了一定基础[14]。然而,目前对红景天基因组SSR的研究依然较少,特别是关于主要入药种大花红景天的SSR位点尚无报道。
本研究基于大花红景天茎/叶混样转录组测序(RNA-Seq)所获得的数据,利用MISA软件对SSR标记进行搜索,分析其分布与组成特征,并对其扩增效率和通用性进行了初步评价,以期为红景天属植物的分类与鉴定、亲缘关系和遗传多样性研究、种质资源利用和分子标记辅助育种提供理论基础。
1 材料与方法
1.1 材料
本研究用于引物筛选和通用性分析的所有植物材料均为野生种,于2015年7月15日至7月22日采自中国西藏地区。4个地理居群的大花红景天(Rhodiola crenualta)分别采集于拉萨市米拉山(RcML)海拔4 868.4 m处、山南地区浪卡子县(RcLK)海拔5 237.2 m处、林芝市色季拉山(RcSJ)海拔4 688 m处、那曲地区嘉黎县(RcJL)海拔4 499 m 处。其他6种红景天属材料分别为长鞭红景天(Rhodiola fastigiata,Rfa)、柴胡红景天(Rhodiola bupleuroides,Rbu)、圣地红景天(Rhodiola sacra,Rsa)、菊叶红景天(Rhodiola chrysanthemifolia,Rch)、狭叶红景天(Rhodiola kirilowii,Rki)和高山红景天(Rhodiola cretinii,Rcr)。将上述材料的叶片用无菌水洗净并擦干后,再用液氮速凍,于-80 ℃保存备用。
1.2 方法
1.2.1 DNA提取和PCR扩增 红景天叶片基因组的提取采用改良的十六烷基三甲基溴化铵(CTAB)法[15]。用1%琼脂糖凝胶电泳和可见分光光度计NanoDrop 1000检测样品DNA的质量,保证所有DNA样品的浓度在300~500 ng/μL之间,D260 nm/D230 nm和D260 nm/D280 nm均在1.8~2.2之间,且无RNA污染。将各DNA样品稀释到浓度为50 ng/μL后于-20 ℃保存备用。
PCR扩增反应体系为25 μL,包括2×U Taq PCR Mix(Zoman Biotechnology,Beijing,China)12.5 μL、上下游引物各1 μL、模板DNA 1 μL、ddH2O 9.5 μL。PCR扩增程序:94 ℃ 2 min;94 ℃ 30 s,56 ℃ 30 s,72 ℃ 30 s,33个循环;72 ℃ 5 min,4 ℃保存。扩增产物用1%琼脂糖凝胶进行电泳检测。
1.2.2 转录组数据来源 以海拔4 868.4、5 237.2 m处的大花红景天(RcML、RcLK)为材料,用CTAB法分别提取叶片、茎的总RNA,等量混匀后,分别标记为Rc4800、Rc5200后备用[16]。转录组测序由上海欧易生物医学科技有限公司完成,使用Illumina HiSeqTM 2500测序仪完成。将测序得到的reads用Trinity(vesion:trinityrnaseq_r20131110)软件paired-end的拼接方法得到Transcript序列,并用TGICL软件延伸得到Unigene[17-18]。
1.2.3 转录组SSR检测和引物设计 用软件MISA(http://pgrc.ipk-gatersleben.de/misa/)对转录组拼接unigene的SSR位点进行搜索。搜索标准如下:重复单元长度为1~6 bp时,单核苷酸重复次数≥10次,二核苷酸重复次数≥6次,三、四、五、六核苷酸重复次数≥5次。
用Primer Premier 3软件对具有SSR位点的unigene序列设计引物。设计原则如下:引物序列长度为18~25 bp,扩增产物大小为80~300 bp,GC含量为40%~65%,退火温度为55~65 ℃且上、下游引物的退火温度相差不大于2 ℃,避免出现错配、引物二聚体和发卡结构。从设计好的引物中随机选择280对送至生工生物工程(上海)股份有限公司合成。
2 结果与分析
2.1 大花红景天转录组测序、unigene组装与注释
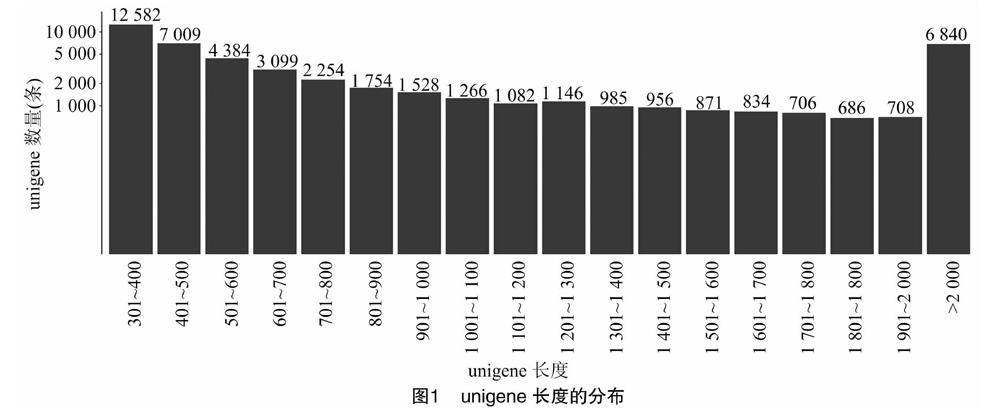
通过Illumine测序平台共得到81 105 580 raw reads(原始读序),通过对raw reads进行质量监控,过滤低质量的reads后,Rc5200获得40 584 060个clean reads(有效读序),Rc4800获得40 521 520个clean reads。用de novo拼接技术将有overlap(重叠区)的clean reads连接成1个更长的序列,使用Trinity (vesion:trinityrnaseq_r20131110)软件的paired-end方法不断延伸然后拼接成transcript(转录本序列),通过TGICL软件聚类、去掉冗余序列,最终得到1套unigene(单基因序列)(图1)。本研究共得到48 790条unigene,总长度为50 851 045 bp,平均大小为1 042.24 bp,其中最长的为15 505 bp,最短的为301 bp。
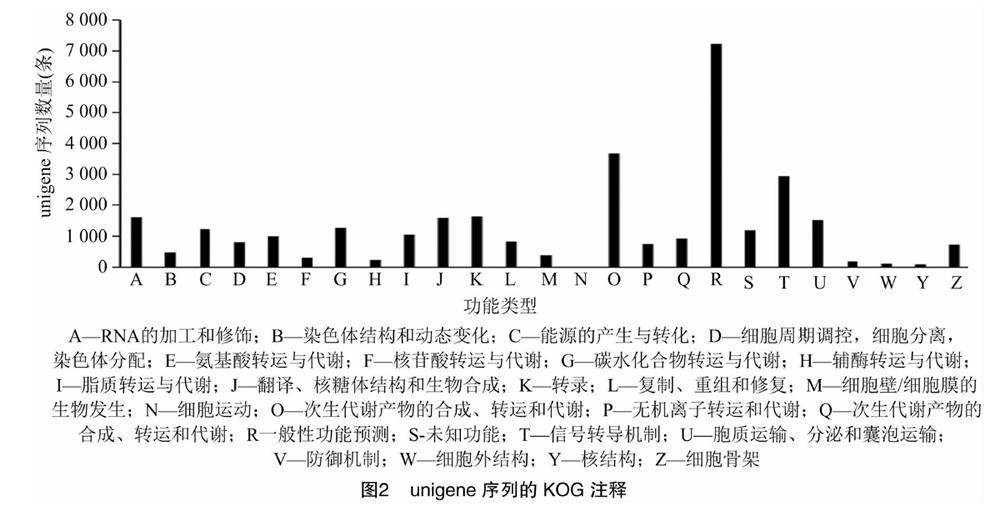
基于BLAST对de novo拼接的unigene进行功能注释和分类,通过BLASTx将unigene序列分别与NR、SWISSPROT和KOG库进行比对,取e<1×10-5的注釋,选择与unigene相似性最高的蛋白,将其功能作为该unigene蛋白功能的注释信息。5个数据库注释统计结果如下:NR注释到31 024条(63.59%),SWISSPROT注释到23 770条(48.72%),KOG注释到19 792条(40.57%),KEGG注释到7 773条(15.93%),GO注释到20 660 条(42.34%)。
根据KOG功能分类的结果,其中大部分unigene注释到R(即一般性功能预测),其数量为7 228个,3 665条unigene注释到O(即次生代谢产物的合成、转运和代谢), 2 942条unigene注释到T(即信号转导机制),此外K(转录)、A(RNA的加工和修饰)、J(翻译、核糖体结构和生物合成)、U(胞质运输、分泌和囊泡运输)、G(碳水化合物转运与代谢)、C(能源的产生与转化)、I(脂质转运与代谢)、E(氨基酸转运与代谢)也均在1 000条unigene以上。GO功能分类可以在基因的分子功能、细胞成分、参与的生物过程3个方面对其进行分类。对所获得的unigene基因进行GO分类,结果显示在分子功能中,binding、catalytic activity(结合、催化活性)类最多;在细胞成分中,cell、cell part(细胞、细胞成分)最多,分别为15 830、15 792条;在生物过程中,cellular process、metabolic process(细胞过程、代谢过程)最多,分别为14 116、12 057条(图2)。
2.2 大花红景天转录组SSR位点的分布
用软件MISA对48 790条unigene的SSR位点进行搜索,结果显示,符合条件的SSR共有10 761个,分布频率(总SSR位点数/总unigene数)为22.06%,平均分布距离(总unigene长度/总SSR位点数)为4.725 kb。含有SSR位点的unigene为8 973 条,发生频率(含有SSR位点的unigene/总unigene数)为18.39%。其中有1 512条unigene含有2个及以上SSR位点,449条unigene含有复合SSR位点。SSR位点的序列总长度为16 590 bp。
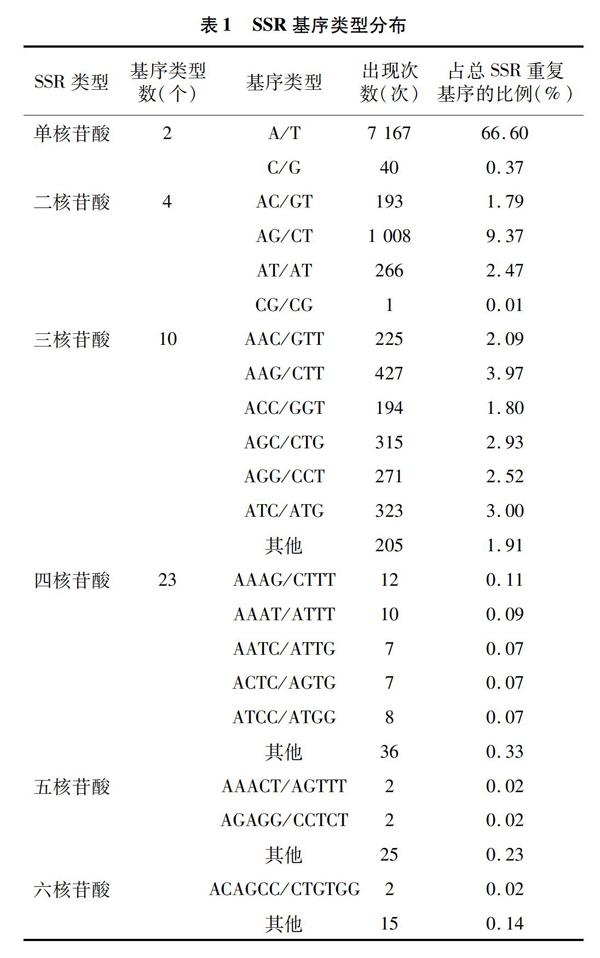
由表1可以看出,大花红景天转录组SSR的类型较为丰富,单核苷酸、多核苷酸重复类型均有分布,但是不同类型的比例差异显著。主要为单核苷酸重复类型,为7 207个,占总SSR位点数的66.97%,平均分布距离为5.622 kb。其次为二核苷酸、三核苷酸重复类型,分别含有1 468、1 960个位点,分别占SSR位点数的13.64%、18.21%,平均分布距离分别为27.603、20.674 kb;四、五、六核苷酸重复类型较少,均不多于80个位点,分别为80、29、17个。
2.3 大花红景天转录组SSR基序重复类型和频率特征
对大花红景天转录组SSR基序类型进行分析显示,共有82种重复基序,单、二、三、四、五、六核苷酸重复各有2、4、10、23、27、16种;重复次数主要为5~24次,不同重复次数的SSR位点数目不同,主要为5、10、11次重复类型(图3)。
由表1可以看出,大花红景天转录组不同SSR基序出现的频率相差较大,以单、二、三核苷酸重复基序为主要类型,占总SSR位点的98.83%。其中以基序A/T最多,为7 167个,其次为AG/CT(1 008个)。单核苷酸重复基序主要为A/T,重复次数集中在10~14次,而G/C较少,仅有40个,且重复次数主要为10~13次。二核苷酸重复基序主要为AG/CT,其次为AT/AT(266个)和AC/GT(193个),CG/CG仅含有1个,主要重复次数为5~11次。三核苷酸重复基序中AAG/CTT最多,为427个,其次为ATC/ATG(323个)、AGC/CTG(315个)、AGG/CCT(271个)、AAC/GTT(225个)、ACC/GGT(194个),其余4种均未超过100个,主要重复次数为5~7次。四、五、六核苷酸重复类型虽然最多,但是数量较少,除AAAG/CTTT、AAAT/ATTT类型分别为12、10个外,其他66种类型均低于10个。
2.4 大花红景天SSR引物的开发和通用性检测
基于含有SSR位点的unigene序列,利用Primer Premier 3软件设计引物,每条unigene产生3对引物。共得到18 696对符合条件的引物,分别位于6 232条unigene上。随机挑选280对引物,包括单核苷酸(125对)、二核苷酸(32对)、三核苷酸(48对)、四核苷酸(51对)、五核苷酸(14对)、六核苷酸(9对)。以4个地理居群的大花红景天DNA为模板对上述引物进行扩增,共246对引物能够扩增出与预期产物大小一致的特异性条带,有效扩增率为87.86%。不同SSR重复类型引物的扩增成功率分别为89.6%、90.63%、79.17%、94.12%、78.57%、88.89%。此外54/246对引物在不同产地大花红景天中的扩增产物具有多态性,占有效引物的21.95%。共扩增得到167条电泳条带,其中多态性片段为95条,平均每对引物产生的多态性片段为1.76条(图4)。
为了进一步验证大花红景天转录组SSR引物是否对其他种具有通用性,在246对引物中随机选择80对引物,以6种红景天(Rfa、Rbu、Rki、Rcr、Rsa、Rch)的DNA为模板进行扩增。结果显示,大花红景天转录组SSR引物对长鞭红景天、狭叶红景天、菊叶红景天、圣地红景天、柴胡红景天和高山红景天的PCR扩增成功率较高,分别为75%、73.75%、72.5%、62.5%、70.0%、72.5%。用筛选到的54对大花红景天SSR多态性引物(表2)对20个样品,包括4个产地(RcML、RcLK、RcJL、RcLZ)的大花红景天和6种红景天进行PCR扩增,每个样品包含2个重复。结果显示,45/54对SSR引物在10个样本中均能够扩增出清晰明亮的多态性产物條带。基于扩增产物多态性信息,利用NtSYS 2.1软件构建上述样品的遗传进化树,结果显示,红景天属植物的种间遗传多态性显著高于种内。由此可见,不同地理居群的大花红景天明显聚为1支,狭叶红景天、菊叶红景天和圣地红景天明显聚为1支,柴胡红景天、长鞭红景天和高山聚为1支(图5)。
3 讨论与结论
近年来,随着红景天药用价值和保健价值的逐渐开发,人们对其需求量也与日俱增。然而由于缺少人工栽培技术,对红景天种质资源的掠夺和破坏十分严重,从而引起多个物种资源数量急剧下降,极大破坏了冻原植被带的高寒草甸生态系统[19]。此外,红景天属植物多分布于海拔较高的山区,因生长环境恶劣,常伴有低温、低氧、强紫外等特点,使得对其采集和研究均十分困难[20-22]。本研究对
2种海拔地区的大花红景天进行转录组测序,基于测序结果分析SSR分子标记的类型与分布情况,并对SSR引物的有效性和通用性进行评估,为进一步开展红景天属植物遗传多样性分析、物种鉴定、遗传图谱构建、分子标记辅助育种和功能基因的发掘提供坚实的理论基础。
根据表达序列标签(EST)开发SSR分子标记具有简单、便捷等特点,特别对于尚无参考基因组序列的非模式植物而言,利用转录组测序技术可以获得大量基因序列,为开发该物种的SSR分子标记提供有力的基础[23]。目前已对多个物种的SSR位点进行了比较详尽的研究,如黑绿豆(Vigna mungo)[24]、花生(Arachis hypogaea)[25]、绿豆(Vigna radiate)[26]、赤小豆(Vigna umbellata)[27]、豇豆(Vigna unguiculata)[28]、洋麻(Hibiscus cannabinus)[29]、开心果(Pistacia vera)[30]、白桦(Betula platyphylla)[31]、苜蓿(Medicago sativa)[32]和刺梨(Rosa roxburghii)[33]等。本研究通过对转录组组装的unigene序列SSR位点进行搜索,共得到10 761 个SSR,分布频率为22.06%,平均分布距离为4.725 kb,与已报道的万寿菊(Tagetes erecta)[34]、柑橘(Citrus sinensis osbeck)[35]、萝卜(Raphanus sativus)[36]等相似。不同物种的SSR重复类型十分不同,如大花红景天SSR重复基序类型以单核苷酸、二核苷酸和三核苷酸重复类型最多,占总位点数的98.83%,且A/T重复10次的最多,为2 899个,占总位点的26.93%,这一结果与绿豆的SSR分布相似[26]。ACG/AGT、CG/CG重复基序比例最少,与刺梨的结果一致[33]。
在随机合成的280对引物中,有249对引物可以在4个地理居群的大花红景天中成功扩增,比例较高,为87.86%,且筛选到54对多态性引物。说明大花红景天的种内遗传变异较小。此外,随机挑选的80对引物对6种红景天植物扩增成功率均在60%以上,说明利用转录组数据开发的大花红景天SSR具有较高的通用性,为其他红景天种材料提供了较为丰富的SSR信息来源。通过对多态性扩增产物构建进化树可以观察到红景天属植物的种内遗传变异明显小于种间,且圣地红景天与菊叶红景天明显聚为一支,长鞭红景天与柴胡红景天聚类关系较近,这与笔者所在实验室前期基于DNA条形码(ITS)所获得红景天属植物聚类结果[37]一致。说明SSR分子标记技术可以作为区分红景天属植物类群的重要手段。
参考文献:
[1]中国科学院中国植物志委员会,等. 中国植物志:景天科[M]. 北京:科学出版社,1984.
[2]Liu Z L,Liu Y Y,Liu C S,et al. The chemotaxonomic classification of Rhodiola plants and its correlation with morphological characteristics and genetic taxonomy[J]. Chemistry Central Journal,2013,7(1):118-126.
[3]Zhang J Q,Meng S Y,Wen J,et al. DNA barcoding of Rhodiola (Crassulaceae):a case study on a group of recently diversified medicinal plants from the Qinghai-Tibetan Plateau[J]. PLoS One,2015,10(3):1-15.
[4]Zhang J Q,Meng S Y,Wen J,et al. Phylogenetic relationships and character evolution of Rhodiola (Crassulaceae) based on nuclear ribosomal ITS and plastid trnL-F and psbA-trnH sequences[J]. Systematic Botany,2014,39(2):441-451.
[5]Kelly G S. Rhodiola rosea:a possible plant adaptogen[J]. Alternative Medicine Review A Journal of Clinical Therapeutic,2001,6(3):293-302.
[6]Xin T Y,Li X J,Yao H,et al. Survey of commercial Rhodiola products revealed species diversity and potential safety issues[J]. Sci Rep,2015,5:8337-8342.
[7]Baran M G,Baranek K B,Pietrosiuk A. Biotechnological approaches to enhance salidroside,rosin and its derivatives production in selected Rhodiola spp. in vitro cultures[J]. Phytochem Rev,2015,14(4):657-674.
[8]國家药典委员会. 中华人民共和国药典:一部[M]. 北京:中国医药科技出版社,2015:154.
[9]Hou Y,Lou A. Population genetic diversity and structure of a naturally isolated plant species,Rhodiola dumulosa (Crassulaceae)[J]. PLoS One,2011,6(9):1-10.
[10]Chen J F,Li R H,Xia Y S,et al. Development of EST-SSR markers in flowering Chinese cabbage (Brassica campestris L. ssp. chinensis var. utilis Tsen et Lee) based on de novo transcriptomic assemblies[J]. PLoS One,2017,12(9):1-14.
[11]Luo H Y,Xu Z J,Li Z D,et al. Development of SSR markers and identification of major quantitative trait loci controlling shelling percentage in cultivated peanut (Arachis hypogaea L.)[J]. Theoretical and Applied Genetics,2017,130(8):1635-1648.
[12]Kempk K,Mora-irtiz M,Ssith L M,et al. Characterization of novel SSR markers in diverse sainfoin (Onobrychis viciifolia) germplasm[J]. BMC Genet,2016,17(1):124-138.
[13]You J,Liu W,Zhao Y,et al. Microsatellite markers in Rhodiola (Crassulaceae),a medicinal herb genus widely used in traditional Chinese medicine[J]. Applications in Plant Sciences,2013,1(3):1-4.
[14]雷淑芸,高庆波,付鹏程,等. 基于Solexa高通量测序的唐古特红景天(Rhodiola algida)微卫星信息分析[J]. 植物研究,2014,34(6):829-834.
[15]赵 为,邓科君,杨足君,等. 景天科植物基因组DNA的高效提取方法[J]. 安徽农业科学,2006,34(22):5804-5805.
[16]张力鹏,张银兴,宋文芹,等. 红景天属植物叶片RNA高效提取的方法[J]. 南开大学学报(自然科学版),2017,50(6):48-53.
[17]Pertea G,Huang X Q,Liang F,et al. TIGR gene indices clustering tools (TGICL):a software system for fast clustering of large EST datasets[J]. Bioinformatics,2003,19(5):651-652.
[18]Grabherr M G,Haas B J,Yassour M,et al. Trinity:reconstructing a full-length transcriptome without a genome from RNA-Seq data[J]. Nature Biotechnology,2011,29(7):644-652.
[19]孟慶文,崔卫东,白光红,等. 红景天种内遗传多样性分析AFLP方法建立[J]. 新疆农业科学,2008,45(1):88-92.
[20]Fu Y,Li L,Hao S,et al. Draft genome sequence of the Tibetan medicinal herb Rhodiola crenulata[J]. Gigascience,2017,6(6):1-5.
[21]Ahemed S,Zhan C S,Yang Y Y,et al. The transcript profile of a traditional Chinese medicine,Atractylodes lancea,revealing its sesquiterpenoid biosynthesis of the major active components[J]. PLoS One,2016,11(3):1-19.
[22]Zhang L P,Wu M,Yu D S,et al. Identification of glutathione peroxidase (GPX) gene family in Rhodiola crenulata and gene expression analysis under stress conditions[J]. International Journal of Molecular Sciences,2018,19(11):3329-3346.
[23]刘 超,张力鹏,王春国,等. 日本落叶松EST-SSR标记挖掘及特征分析[J]. 林业科学研究,2013,26(f10):60-68.
[24]Souframanien J,Reddy K S,Denovo A. De novo assembly,characterization of immature seed transcriptome and development of genic-SSR markers in black gram Vigna mungo L. Hepper[J]. PLoS One,2015,10(6):1-18.
[25]Bosamia T C,Mishra G P,Thankappan R,et al. Novel and stress relevant EST derived SSR markers developed and validated in peanut[J]. PLoS One,2015,10(6):1-19.
[26]Chen H L,Wang L X,Wang S H,et al. Transcriptome sequencing of mung bean (Vigna radiate L.) genes and the identification of EST-SSR markers[J]. PLoS One,2015,10(4):1-15.
[27]Chen H L,Chen X,Tian J,et al. Development of gene-based SSR markers in rice bean (Vigna umbellata L.) based on transcriptome data[J]. PLoS One,2016,11(3):1-13.
[28]Chen H L,Wang L X,Liu X Y,et al. De novo transcriptomic analysis of cowpea (Vigna unguiculata L. Walp.) for genic SSR marker development[J]. BMC Genet,2017,18(1):65-77.
[29]Li H,Li D,Chen A,et al. Characterization of the kenaf (Hibiscus cannabinus) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers[J]. PLoS One,2016,11(3):1-8.
[30]Ziya M E,Kafkas S,Khodaeiaminjan M,et al. Genome survey of pistachio (Pistacia vera L.) by next generation sequencing:Development of novel SSR markers and genetic diversity in Pistacia species[J]. BMC Genomics,2016,17(1):998-1012.
[31]Hao W,Wang S J,Liu H J,et al. Development of SSR markers and genetic diversity in white birch (Betula platyphylla)[J]. PLoS One,2015,10(4):1-14.
[32]Wang Z,Yu G H,Shi B B,et al. Development and characterization of simple sequence repeat (SSR) markers based on RNA-sequencing of Medicago sativa and in silico mapping onto the M. truncatula genome[J]. PLoS One,2014,9(3):1-7.
[33]Yan X Q,Zang X,Lu M,et al. De novo sequencing analysis of the Rosa roxburghii fruit transcriptome reveals putative ascorbate biosynthetic genes and EST-SSR markers[J]. Gene,2015,561(1):54-62.
[34]張华丽,丛日晨,王茂良,等. 基于万寿菊转录组测序的SSR标记开发[J]. 园艺学报,2018,45(1):159-167.
[35]Jiang D,Zhong G Y,Hong Q B. Analysis of microsatellites in Citrus unigenes[J]. Acta Genetica Sinica,2006,33(4):345-353.
[36]Wang S F,Wang X X,He Q W,et al. Transcriptome analysis of the roots at early and late seedling stages using Illumina paired-end sequencing and development of EST-SSR markers in radish[J]. Plant Cell Reports,2012,31(8):1437-1447.
[37]张力鹏,滕艳娇,于得水,等. 西藏地区红景天属植物ITS、rbcL、trnS-G序列多态性分析[J]. 南开大学学报(自然科学版),2019,52(5):93-101.
猜你喜欢
江苏农业科学(2017年18期)2017-11-18
江苏农业科学(2017年18期)2017-11-18
科技创新导报(2017年19期)2017-09-13
中国中药杂志(2017年15期)2017-08-30
中国中药杂志(2017年15期)2017-08-30
中国中药杂志(2017年13期)2017-07-31
中国中药杂志(2017年4期)2017-03-28
中国中药杂志(2017年2期)2017-03-25
中国中药杂志(2017年1期)2017-03-06
