
NLRP3对病毒感染心肌细胞凋亡及炎症反应的作用*
2021-07-26 03:13李卫东颜源均赵世桥冯尧蒲志强冯飞
西部医学 2021年7期
李卫东 颜源均 赵世桥 冯尧 蒲志强 冯飞
(川北医学院附属医院急诊医学科,四川 南充 637000)
病毒性心肌炎(viral myocarditis,VMC)是由嗜心肌病毒感染心肌形成以心肌严重的炎症反应导致心肌细胞损伤为主的心脏疾病。常见病毒为柯萨奇病毒B组(coxsackie virus B,CVB)。病毒性心肌炎多发生于青少年,大部分病情较轻,能够痊愈;但有少部分病情较重而导致患者死亡,还有一部分发展成为扩张型心肌病。研究[1]报道,炎症反应及心肌细胞凋亡是导致病毒性心肌炎早期心肌细胞损伤的主要机制。在病毒性心肌炎炎症反应中,炎症趋化因子IL-1β、IL-18在其中起到重要作用,IL-1β、IL-18在机体内是以无功能的前体形式存在的,NLRP3能够通过凋亡相关斑点样蛋白ASC(apoptosis-associated speck-like protein containing a CARD)寡集半胱天冬蛋白酶的前体(procaspase-1)从而激活门冬氨酸特异的胱氨酸蛋白酶-1(cysteinyl aspartate specific proteinase,caspase-1),激活的caspase-1能对pro-IL-1β、pro-IL-18进行加工切割,形成成熟的具有功能的IL-1β、IL-18并分泌到细胞外发挥作用[2]。因此,本研究探讨核苷酸结合寡聚化结构域样受体蛋白3(nucleotide-binding oligomerization domain-like receptor protein 3,NLRP3)炎性体在病毒感染早期心肌细胞的作用以及与心肌细胞凋亡之间的关系,为寻找病毒性心肌炎有效的治疗靶点奠定基础。
1 材料和方法
1.1 材料
1.1.1 病毒和实验动物 CVB3(Nancy株)购于美国典型培养物保藏中心(ATCC)。出生1~2 d的SPF级Sprague-Dawley(SD)大鼠乳鼠由川北医学院实验动物中心提供。
1.1.2 主要试剂及仪器 抗NLRP3、ASC及HRP-结合次级产物的抗体,检测IL-β、IL-18、CK-MB、cTnI等的ELISA试剂盒,琼脂糖购自BOSTER公司,胰蛋白酶购于Hyclone,BCA蛋白浓度测定试剂盒、RIPA细胞裂解液、annexin V-FITC/PI 细胞凋亡检测试剂盒购于碧云天生物科技有限公司,逆转录试剂盒、SYBR荧光定量RT-PCR试剂盒购自TaKaRa公司,Lipofectamine 2000试剂购于Invitrogen,NLRP3 siRNA购于Santa。
1.2 方法
1.2.1 原代心肌细胞培养 根据既往的文献[3]报道采用差速贴壁方法分离培养原代心肌细胞。于无菌条件下迅速获取出乳鼠心脏,将获取的心脏组织剪切成1~2 mm3的小块组织并装入含有约10 mL 0.1%胰蛋白酶的三角瓶中,反复消化后加入含血清的培养液终止消化,以4000 r/min离心约10 min,而后弃去上清液。于残留的固体中加入含有10%FBS的DMEM培养液中混匀。于37 ℃的含有5% CO2的培养箱中贴壁1.5~2 h,吸出含有未贴壁细胞的培养液,于显微镜下计数完成后调整浓度为3×105/mL接种于含有5-溴脱氧尿苷(最终浓度为0.1 mol/L,抑制非心肌细胞生长)与10% FBS的DMEM培养液中,置于5%CO2培养箱中 37 ℃继续培养2~3 d,显微镜下可观察到85%的细胞为心肌细胞且具有明显的搏动。
1.2.2 NLRP3基因沉默及CVB3感染心肌细胞模型的建立 设计NLRP3的小干扰RNA(siRNA)以转染至培养好的SD大鼠心肌细胞中,具体的siRNA转染操作参照美国Invitrogen公司转染试剂盒中推荐的转染步骤,具体步骤如下:转染前利用PBS将培养48 h的原代心脏细胞洗涤2次,以5×103细胞/孔接种于6孔板中。将siRNA溶液与lipofectamin2000溶液混合液1 mL/孔,分别加入细胞培养孔,孵育6 h,换液继续培养24 h后完成转染。将CVB3 Nancy株接种在HeLa细胞上并测定50%组织感染率(50% tissue culture infective does,TCID50),选100 TCID50感染心肌细胞。在心肌细胞NLRP3基因沉默后的第二天加入100 TCID5CVB3病毒液100 μL于37 ℃含有5% CO2的恒温培养箱中培养2 h更换为普通培养液,得到CVB3诱导的心肌炎心肌细胞模型。实验包含4个组别,分别包括正常对照组、Scrambled-siRNA组、CVB3+Scrambled-siRNA组、CVB3+NLRP3-siRNA组。
1.2.3 ELISA检测各组IL-1β、IL-18、CK-MB、cTnI的水平 各实验组心肌细胞处理24 h后,按照ELISA试剂盒说明书进行检测。以标准品的OD值为横坐标,浓度为纵坐标,绘制标准曲线,得到标准曲线回归方程式,将样品的OD值带入方程式,计算出样品浓度,再乘以稀释倍数即得样品实际浓度。
1.2.4 RT-qPCR检测各组中NLRP3的mRNA表达 严格按照RT-qPCR试剂盒说明书进行检测。NLRP3 的上游引物序列为5′-ACGGCAAGTTCGA AAAAGGC-3′,下游引物序列为 5′-AGACCTCGG CAGAAGCTAGA-3′,扩增产物长度为87 bp;β-actin 的上游引物序列为5′-AGACAGCCGCATCTTCTT GT-3′,下游引物序列为5′-TGATGGCAACAAT GTCCACT-3′,扩增产物长度为142 bp。严格按照SYBR®Premix Ex Taq II(Tli RNaseH Plus)说明书操作,整个过程中在加入ROX后需避光操作,冰上操作。由软件记录CT值,计算2-ΔΔCT值作为mRNA的表达水平。
1.2.5 Western blot检测各组中NLRP3、caspase-1、IL-1β、cleaved caspase 1、cleaved caspase-3、caspase-8、cleaved caspase-9、Bcl-2、bax的蛋白水平 各实验组心肌细胞处理24 h后,按照说明书提取各组总蛋白。采用BCA蛋白含量检测试剂盒检测各处理组细胞中的总蛋白量,SDS-PAGE凝胶电泳(每孔上样100 μg),电泳结束后将蛋白转移至PVDF膜,5%脱脂奶粉封闭后,分别加入抗NLRP3、caspase-1、IL-1β、cleaved caspase-1、cleaved caspase-3、caspase-8、cleaved caspase-9、Bcl-2、bax抗体稀释液(1∶400)、GAPDH内参稀释液(1∶1000),4 ℃孵育过夜,第二天加入封闭液稀释的Ⅱ抗(1∶4000)。充分漂洗后以ECL法显像,将膜放入曝光仪中曝光,根据条带显色的强弱选择曝光时间,Image J软件统计各图灰度值,计算蛋白的相对表达量。

2 结果
2.1 CVB3病毒感染心肌细胞中NLRP3基因及蛋白表达情况以及NLRP3基因沉默 RT-qPCR实验结果表明,CVB3感染后NLRP3的mRNA表达量明显升高在第12 h达到高峰,后其表达量稍有下降并趋于稳定,随着培养时间的延长其表达量逐渐降低(图1A)。Western blot检测结果表明,CVB3感染后NLRP3的蛋白表达量明显升高且24 h达到高峰,随后开始降低,这与NLRP3的mRNA升高基本一致(图1B)。在敲减NLRP3基因表达后,CVB3感染心肌细胞后RT-qPCR及Western blot检测结果表明,敲减NLRP3基因的表达可以明显降低病毒感染心肌细胞中NLRP3基因及蛋白的表达(P<0.05)(图1C、D),表明用基因沉默技术敲减NLRP3表达是成功的。
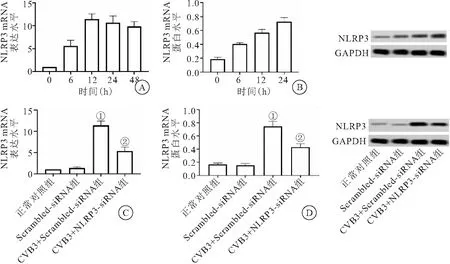
图1 CVB3病毒感染心肌细胞NLRP3基因及蛋白表达的情况
2.2 敲减NLRP3基因的表达可一定程度减轻心肌细胞损伤 ELISA方法检测培养上清液中心肌损伤标志物CK-MB、cTnI结果表明,与正常对照组比较,CVB3+Scrambled-siRNA组明显升高(P<0.01),敲减NLRP3表达后,同CVB3+NLRP3-siRNA组比较,CK-MB、cTnI的表达均有不同程度降低(P<0.01),见图2。

图2 ELISA检测各组中CK-MB、cTnI的水平
2.3 敲减NLRP3基因的表达对病毒感染心肌细胞中NLRP3下游因子caspase-1、IL-1β、IL-18的影响 在心肌细胞感染CVB3病毒的6~24 h,caspase-1的蛋白表达量明显增加,见图3A。敲减乳鼠心肌细胞中NLRP3基因的表达后,cleaved caspase-1的蛋白水平明显降低,与CVB3+Scrambled-siRNA组相比差异有统计学意义(P<0.05),见图3B。与CVB3+Scrambled-siRNA组相比,敲减NLRP3基因的表达后IL-1β、IL-18的含量明显下降(P<0.05),见图3C、D。
同时采用Western blot检测敲减NLRP3基因的表达后CVB3感染24 h IL-1β蛋白的变化,与CVB3+Scrambled-siRNA组相比较,敲减NLRP3基因的表达后IL-1β的蛋白水平明显降低,差异具有统计学意义(P<0.01),见图3E。与ELISA检测结果一致。进一步证明了抑制NLRP3的激活,可以抑制CVB3感染心肌细胞导致的IL-1β、IL-18的增加。

图3 NLRP3基因沉默后其下游因子caspase-1、IL-1β、IL-18蛋白水平的变化
2.4 敲减NLRP3基因的表达对病毒感染心肌细胞凋亡相关蛋白的影响 正常对照组比较,CVB3+Scrambled-siRNA组的caspase-8表达明显升高,差异具有统计学意义(P<0.05);但敲减NLRP3基因的表达后,与CVB3组比较caspase-8表达水平的差异无统计学显著性(P>0.05)。与正常对照组相比,CVB3+Scrambled-siRNA组cleaved caspase-3的蛋白水平明显升高,差异有统计学意义(P<0.05);敲减NLRP3基因的表达后,cleaved caspase-3的蛋白水平与CVB3+Scrambled-siRNA组比较明显降低(P<0.05);Western blot法检测cleaved caspase-9的结果表明,CVB3+Scrambled-siRNA组的cleaved caspase -9明显高于正常对照组(P<0.05);敲减NLRP3基因的表达后,与CVB3+Scrambled-siRNA组相比较,cleavedcaspase-9的蛋白水平有所下降(P<0.05),见图4。

图4 各组心肌细胞中凋亡蛋白caspase-8、cleaved caspase-3、cleaved caspase-9蛋白水平的变化
2.5 敲减NLRP3基因的表达对CVB3病毒感染心肌细胞中Bcl-2、Bax的影响 Western blot检测与线粒体凋亡途径相关的Bcl-2与Bax蛋白表达情况:与正常对照组相比,CVB3+Scrambled-siRNA组的Bcl-2蛋白表达量降低及Bax蛋白的表达明显增加,Bax/Bcl-2比值增高(P<0.05)。敲减NLRP3基因的表达后,与CVB3+Scrambled-siRNA组对比, Bcl-2蛋白降低幅度减弱(P<0.05);但对Bax蛋白表达无明显影响(P>0.05),Bax/Bcl-2比值一定程度降低(P<0.05),见图5。
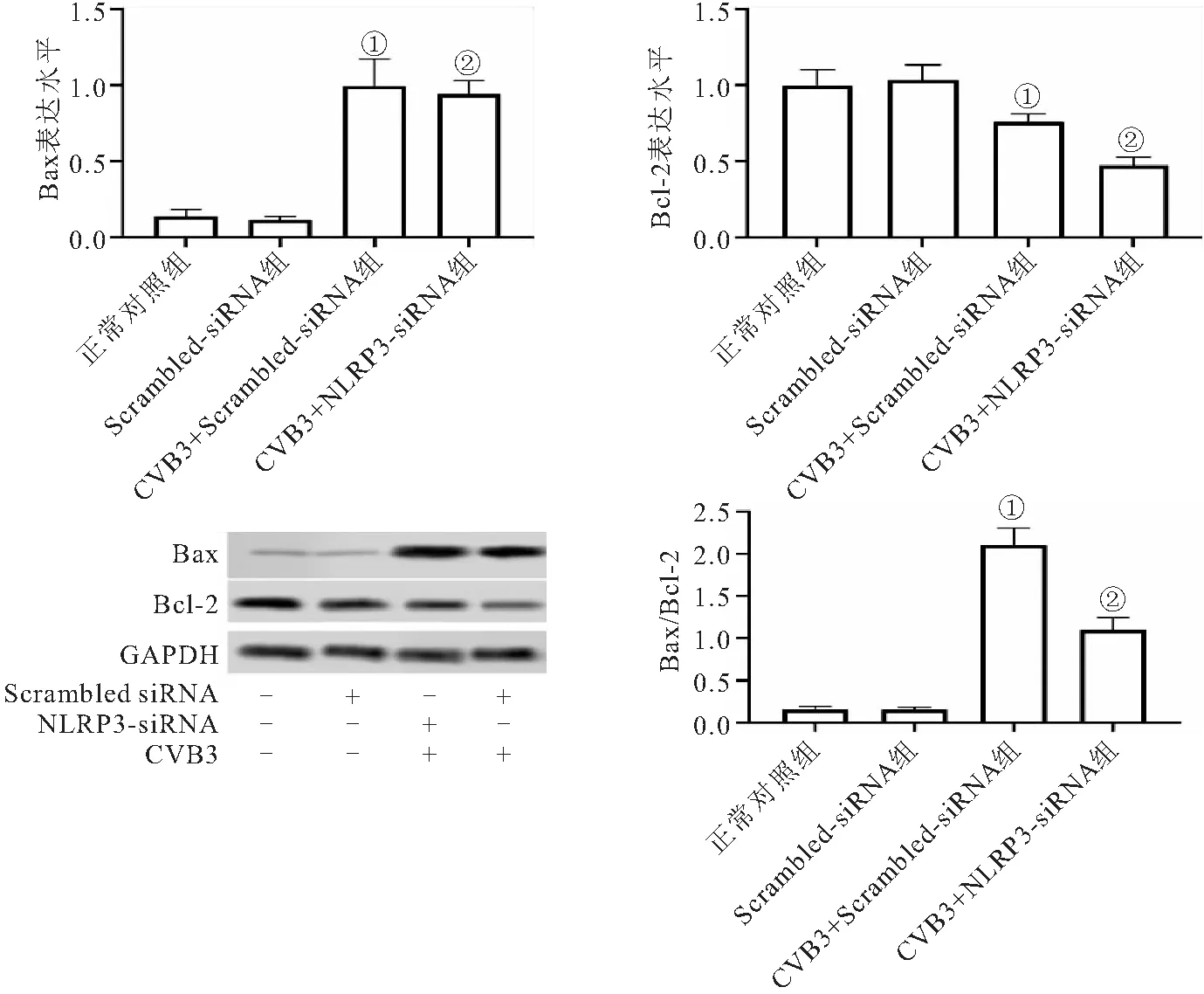
图5 各组心肌细胞凋亡相关蛋白Bcl-2、Bax表达水平的变化
3 讨论
病毒性心肌炎中最常见的病毒感染为CVB3病毒,其对心肌细胞的直接损伤主要包括炎症反应及心肌细胞凋亡[3-4]。病毒感染心肌细胞后激活心肌细胞内相关炎症信号通路,产生IL-18、肿瘤坏死因子-α(TNF-α)、IL-1β、IL-6等炎症趋化因子募集炎症细胞聚集产生炎症反应导致心肌细胞损伤。
NLRP3是NLRP亚家族中的一员,NLRP亚家族包含有NLRP1、NLRP2、NLRP3等,广泛存在于细胞浆中[5]。NLRP3炎性体除了存在于巨噬细胞、中性粒细胞等炎症细胞外,还存在于心肌细胞、血管内皮细胞以及心肌成纤维细胞的胞质中[6]。本课题组前期研究显示,血管紧张素Ⅱ刺激体外培养的H9C2细胞内NLRP3、ASC、Caspase-1和IL-1β的mRNA及蛋白水平表达明显升高,在细胞培养液上清液中IL-1β的含量也升高[7]。抑制心肌细胞中NLRP3的激活可以减轻糖尿病大鼠心肌炎症反应及心肌细胞损伤[8]。病毒性心肌炎导致心肌细胞中NLRP3激活的机制包括病毒RNA介导的激活、细胞内ROS介导的激活和病毒孔蛋白介导的激活等3方面[9-11]。本课题组在病毒性心肌炎动物模型中研究显示,在病毒感染小鼠第3天心肌组织中NLRP3炎性体被激活,其下游的caspase-1、IL-1β、IL-18表达增加,心肌组织中出现炎性细胞聚集[12]。既往研究还显示,在病毒性心肌炎患者外周血中NLRP3被激活,并与疾病的严重程度成正相关,使用参麦及黄芪等药物可以明显抑制NLRP3升高,减轻炎症反应[13-15]。本研究显示,病毒感染的心肌细胞内NLRP3被激活,其下游因子caspase-1表达上调,IL-1β、IL-18生成增多,敲减NLRP3基因的表达后可一定程度抑制CVB3病毒感染引起的心肌细胞中 caspase-1的活化,一定程度抑制IL-1β、IL-18的表达。提示NLRP3参与了病毒感染心肌细胞早期的炎症反应过程,抑制NLRP3的过度激活,有可能减轻病毒性心肌炎的炎症反应损伤。
心肌细胞凋亡是病毒性心肌炎直接损伤心肌细胞的主要机制之一。在病毒性心肌炎中Fas/FasL外源性凋亡途径[16-17],Bcl-2/Bax线粒体凋亡途径均被激活,最终导致caspase-3活化导致细胞凋亡[18-19]。既往研究报道,线粒体损伤可以激活NLRP3,同时NLRP3的激活进一步加重线粒体损伤[20]。在晚期糖基化终末产物诱导的心肌细胞损伤中,NLRP3、caspase-3和caspase-9的表增高,沉默NLRP3可通过抑制NF-κB P65来抑制caspase-3和caspase-9的增加,减轻心肌细胞凋亡[21]。有研究表明利用siRNA敲减NLRP3的表达可抑制心肌缺血再灌注心肌血管内皮细胞凋亡,其机制可能与降低NF-κB的活化,上调抗凋亡蛋白 Bcl-2的表达有关[22]。还有研究显示在高糖培养的肾小管上皮细胞中用siRNA沉默NLRP3可抑制caspase-3的表达而减轻细胞凋亡[23]。shRNA干扰NLRP3后可以减轻心肌缺血再灌注大鼠模型中的心肌细胞损伤及凋亡,降低caspase-9、caspase-3凋亡蛋白的表达,其可能机制与抑制TGF-β/NF-κβp65/TNF-α炎症通路活化有关[24]。敲减NLRP3基因的表达可以改善2型糖尿病大鼠模型中心功能,抑制心肌细胞凋亡[25]。细胞凋亡是一个复杂的过程,多种细胞因子参与其中并受多种基因的调控。在这复杂的多因素调控网络中,除了上述的caspase家族以外,Bcl-2家族在细胞凋亡的过程中也起着重要的作用。因此,本研究为了探讨在病毒感染心肌细胞中NLRP3对心肌细胞凋亡途径的影响,使用siRNA沉默NLRP3基因,研究其对细胞内外凋亡途径的影响。结果发现在CVB3感染的心肌细胞模型中,caspase-8、cleaved caspase-3、cleaved caspase-9的蛋白水平均增加,Bcl-2表达降低,Bax表达增高,Bax/Bcl-2比值增加,细胞凋亡增加。这与既往的研究结果一致[26-27]。当敲减NLRP3基因表达后,cleaved caspase-3、cleaved-caspase-9的蛋白水平降低,Bcl-2降低程度减弱,但是对caspase-8、Bax无明显影响,Bax/Bcl-2比值降低,心肌细胞凋亡降低,提示在病毒性心肌炎中NLRP3可一定程度上促进线粒体凋亡途径,加速细胞凋亡。本文主要在体外病毒感染心肌细胞模型中考察NLRP3的作用,同时只使用了基因干扰技术敲减NLRP3基因的表达来研究NLRP3的作用,未进行NLRP3基因过表达的研究,具有一定的局限性。
4 结论
在病毒感染心肌细胞中NLRP3被激活,其下游的因子表达增高,心肌细胞凋亡增加。抑制NLRP3过度激活,可一定程度上减轻心肌细胞炎症损伤及线粒体途径介导的心肌细胞凋亡,提示NLRP3可能是病毒性心肌炎炎症反应与心肌细胞凋亡相互联系的关键环节。
猜你喜欢
中老年保健(2022年1期)2022-08-17
湖南饲料(2021年4期)2021-10-13
华声文萃(2021年6期)2021-08-25
文萃报·周二版(2021年11期)2021-04-06
小读者之友(2020年4期)2020-05-15
家庭用药(2017年3期)2017-04-17
妇女生活(2016年1期)2016-01-14
中国民族民间医药·下半月(2014年2期)2014-09-26
为了孩子(孕0~3岁)(2001年3期)2001-06-13
为了孩子(孕0~3岁)(2000年10期)2000-06-13
