
酶工程:从人工设计到人工智能
2021-07-24 08:58王雅丽付友思陈俊宏黄佳城廖浪星张永辉方柏山
化工学报 2021年7期
王雅丽,付友思,陈俊宏,黄佳城,廖浪星,张永辉,方柏山,2,3
(1 厦门大学化学化工学院,福建厦门361005; 2 厦门市合成生物学重点实验室,福建厦门361005; 3 福建省化学生物学重点实验室,福建厦门361005; 4 集美大学食品与生物工程学院,福建厦门361021)
引 言
1878 年,Kuhne 首次提出了“酶(enzyme)”的概念。随后,1894 年,Fischer 给出了酶催化“lock and key”模型假设[1],后于1902 年获得诺贝尔化学奖。Sumner 是首个发现酶可以被结晶的科学家,他于1926 年成功分离和结晶了第一个酶——脲酶[2],胃蛋白酶[3]、烟草花叶病毒核蛋白[4]的高纯度蛋白晶体紧随其后分别被Northrop 和Stanley 制备获得,三人共同获得了1946 年的诺贝尔化学奖。晶体结构解析上的突破不仅让人们对酶的分子结构有了初步的认识,并很快被应用于辅助蛋白序列鉴定。Sanger 在前期研究基础上,于1952 年成功报道了胰岛素A 链和B 链的完整氨基酸序列[5-6],并于1958 荣获诺贝尔化学奖。
随着越来越多的天然酶被分离纯化和结晶鉴定[7-8],科学家们又提出了诱导嵌合(induced-fit)[9-10]和keyhole-lock-key 模型[11],进而更好地解释了不同催化域的酶催化过程。酶氨基酸序列与功能之间的关系也开始被关注。定向进化[12]、随机突变[13]、定点突变[14]、同源重组[15]等方法被应用到了酶工程中,并获得了很多表现优异的生物催化剂应用在化学和药物生物合成、生物传感器、食品生产、生物降解等领域[16-17]。
早在20世纪末期,计算机便被应用到了酶的预测改造和设计中[18]。Karplus, Levitt 和Warshel 三位教授,因其在复杂化学体系多尺度模型计算上作出的贡献获得了2013 年的诺贝尔化学奖。Arnold 凭借在生物酶的定向进化上作出的贡献,2018 年斩获此奖。Baker 开发了蛋白结构从头预测工具Rosetta[19],开启了蛋白从头设计的时代[20-21]。最近,Baker因其开发的蛋白从头设计工具,荣获了2021年生命科学重大突破奖(Breakthrough Prize in Life Sciences)。
用计算机设计酶,主要围绕着酶的催化活性、结构稳定性、底物选择性和可溶性表达以及从头设计等方面展开[22-23]。早在70 多年前,Pauling[24]就提出天然酶的改造需求,由于蛋白序列空间的开阔性和对蛋白序列与结构之间关系认识的局限性,计算机酶改造工程一直在与时俱进[25]。本文对不同的人工设计酶的设计方法和分子力场、再设计和从头设计在酶的设计中的相关成果,以及人工智能在酶设计中的方法与案例进行了综述。
1 人工设计酶的关键因素
1.1 分子力场
分子力场的核心是用来描述分子结构与系统能量之间关系的势能公式组合。力场(force field)可以用来计算分子间和分子内的能量,计算酶和底物之间的亲和力,预测侧链的位置[26]。一个经典的经验力场(empirical force field)能量公式集合了键能、键角、二面角、非平面二面角、电荷分布涉及的能量。经典模型和其微调模型可以在QM/MM 计算[27]中较为准确地描述生物分子结构。目前大多数在使用的生物分子力场例如GROMOS[28]、CHARMM[29]、AMBER[30]和OPLS[31]等都主要是使用这些子项来计算能量。Rosetta 是一种基于知识的(knowledge-based)力场[32],其中加入了重新统计获得的势能和通过实验获得的数据作为额外的势能项。Alford 等[33]对最新的Rosetta 能量函数(REF15)做出了详细的介绍。
根据对原子定义的范围,力场又分为全原子力场和联合原子力场,其中全原子力场里定义了所有的原子,而联合原子力场则只定义了重原子和极性氢原子,例如CHARMM、AMBER 和Rosetta 均是全原子力场,GROMOS 则是联合原子力场,而OPLS 力场则包含有全原子和联合原子两个版本。Mackerell[26]是CHARMM 力场的开发者之一,他对生物大分子经验力场做了详细的介绍和讨论。
要实现酶的精准设计,需要能够准确地设计活性位点的侧链构象、柔性结构如loop 构象以及反应过渡态中原子方位朝向,还要计算静电相互作用、分子与溶剂之间的相互作用和熵损失[21]等。力场函数的准确性对酶的设计具有至关重要的作用。精确地描述每个原子的成键和位置是科学家向往的理想状态。时至今日,并没有一个能够100%准确描述分子实际能量的力场,很多力场开发工作者也正在为提高力场的准确性而共同努力[34-36]。
1.2 设计酶的方法

图1 RosettaDesign和IPRO酶的再设计流程[22]Fig.1 The enzyme redesign computational workflow of RosettaDesign and IPRO [22]
Anfinsen[37]提出的热力学假设——蛋白会朝着能量最低的方向去折叠是计算机设计酶遵循的基本法则。总的说来,酶的计算设计与筛选过程即是一个不断向系统能量更低的折叠方式进行搜索的过程[38]。
常 用 全 原 子 模 型 设 计 软 件 包 有RosettaDesign[39]、PoreDesigner[40]、IPRO[41]等。图1 是以RosettaDesign 和IPRO 为例的一个酶的再设计流程图[22],步骤依次为:将底物对接进酶(dock)、固定催化活性位点氨基酸(ensure)、鉴定出底物结合位点氨基酸(substrate)、调整附近氨基酸残基的侧链位置(adjacent),设计序列(design)、能量最小化(energy minimization)、给设计的酶打分排序(rank)。RosettaDesign 和IPRO 的区别在于,前者在序列设计时利用的是蒙特卡罗法随机模型,IPRO 则利用了MILP(mixed-integer linear optimization)定向采样,是一个蛋白设计和优化迭代的过程[41]。
酶的再设计相比酶的从头设计成功率更高。换而言之,酶的从头设计的流程更为复杂、难度更大。新酶的从头设计的流程图如图2[42]。在设计之前,需要研究者对反应的过渡态和最简化的活性位点有一定的认识,这几个活性位点一般被称为“theozyme”,这个活性位点的分布要求在底物周边且有足够的空间来激发反应。由于反应的过渡态实际只发生在短短的几个飞秒之内[43-44],这个过渡态一般以已知晶体结构酶的过渡态类似物作为出发点,或者通过QM 计算来合理分布这个简化模型的过渡态的位置。确定了中心活性位点的位置后,接下来利用RosettaMatch 模块尝试将这个最简化的活性位点置于合适的骨架里[45],然后进行多个循环的序列设计和蛋白优化,对获得的设计进行排序打分,最后挑选高分的设计进行实验验证。
Baker[21]认为计算机从头设计酶的成功率低有三个原因:①模拟催化活性位点的模型并不准确;②设计的酶在催化位点上的位置与实际的有偏差;③在活性位点设计正确的情况下,也会由于周围氨基酸的长程静电和动力学的问题使得催化效率不高或无法进行[21]。Baker 等[33]不断地优化势能函数、累积设计与实验的经验尝试来解决这些难题。最近,Baker 等[46]开发一种基于氢键网络的计算方法设计蛋白催化中心的网络连接,此方法不仅将过渡态的作用网络考虑在内,也将催化位点周围的长程氢键网络考虑进来,有望成为设计整装(fullypreorganized)酶催化剂的起点。
1.3 筛选评估方法
利用计算机设计酶的优势在于能够提供数量巨大的变体库,而如何快速又准确地从库中筛选目标样本是一个重要的科学问题[47]。随着X 射线晶体学[48]、核磁共振的发展[49]和冷冻电子显微镜(cryo-EM)进入原子分辨率时代[50],PDB数据库[51]里已经积累了大量高分辨率蛋白三维结构信息。通过计算机学习现有的数据库里序列与结构之间的关系,有许多算法被开发出来,从不同的角度来筛选目标蛋白。

图2 新酶的从头设计流程示意图[42]Fig.2 Schematic representation of the de novo design of new enzyme [42]
SCHEMA 是一个可用于结构域重组的打分函数,在替换亲本蛋白的同源序列元素时,能够按蛋白结构的最小破坏度去划分区块[52]。SCHEMA 重组算法在酶的重组型改造中能够帮助预测酶和突变库的结构稳定性[53],提升序列多样性、进而超越亲本的性质[54]。Arnold等[55]在此基础上,开发出非连续序列元素的重组方法。
Straub 等[56]利用学习已经解析的蛋白晶体结构里不同氨基酸的侧链的位置和方向,发展了一种分析势能,来预测给定蛋白序列的三维折叠构象。Lin等[57]开发的分析势能学习的是亲缘生物的同源蛋白多序列比对的共进化信息,该打分系统能够评估一个计算获得的结构和已知结构在折叠中的一致性。DrugScore[47]是一个学习了159个酶与配体复合体结构得到的势能公式,它可以通过熵值贡献度等因素来预测和评估配体在酶活性中心的构象。ABACUS2 是Liu 等[58]开发的基于骨架的蛋白结构序列筛选设计工具。TMFoldRec[59]的重点则是预测跨膜分子的折叠。除了上述这些基于学习已有结构开发的分析势能之外,Rosetta3[60-61]、Osprey[62]、Tinker 8[63]、TransCent[64]和IPRO[41]则是将构象搜索算法和打分算法等融合起来的多功能工具包。此外,还有一些优化模型例如OptGraft[65]和OptZyme[66],前者的思想是将结合位点转移到已知的骨架上进行酶的改造,后者是利用活性位点过渡态类似物进行酶的再设计。
在实验验证中,计算机设计的蛋白和酶,实验验证时常存在错误折叠和聚集。Fleishman 等[67]针对性地开发一个开源的基于结构和序列的算法能够提高计算机设计的蛋白异源表达溶解性和稳定性。Goldenzweig等[68]对计算机设计里蛋白稳定性的研究原理的应用做了详细的阐述。
自然界中的天然酶都是经过漫长的进化而来,其催化位点附近存在有相当复杂相互作用网络,有不同带电量的氨基酸巧妙布局来提高质子传递效率,这也正是计算机设计的酶成功率低,初始催化活性低的原因。通过学习已知的晶体结构提升筛选算法的准确性、借助人工智能提升蛋白结构预测的准确性,将会大大缩短酶的设计中耗时耗力的实验室筛选优化过程,能够提高未来酶设计的速度和精确度。
2 人工设计酶的案例
近些年来,科学家们已经在计算机蛋白设计里做出了很多努力,积累了很多成功的案例。通过计算机设计能提升酶的活性[69-70],改变辅酶特异性[71-73]、底物特异性[74]和立体选择性[75],研究蛋白间的相互作用设计蛋白抗体[76-77],设计大分子的蛋白自组装复合体[78-81],设计具有新功能的酶[82-85]。
2.1 酶的再设计
酶的再设计是在天然酶的基础上,利用计算机进行再改造的过程。
Ehren 等[86]基于序列和结构信息组合递归诱变和机器学习的方法,使Sphingomonas capsulata来源的脯氨酸内切酶(PEP)在模拟胃液条件下对胃蛋白酶的抵抗性增加了200 倍,PEP 的绝对酶活提升了20%。
Khoury 等[71]在前期的实验中,通过突变实验改变了木糖还原酶的辅酶特异性,在此基础上利用一个线性规划算法综合范德华力、静电力、溶剂效应来描述辅酶与辅酶结合能大小,提高酶与NADH 的结合能的同时降低其与NADPH 的结合能。最终对8000 个可能突变体进行采样后,获得10 个辅酶NADH 亲和性提升的酶,其中有8 个酶的NADPH 依赖性降低了90%以上。
Grisewood 等[74]同样基于结构指导突变,利用优化算法筛选具有潜力的突变体,最终特异性地提升了Acyl-ACP 硫酯酶水解中链(C8~C12) 脂肪酸的选择性。Wijma 等[75]利用计算机设计催化位点,构建了一个小样本突变库,计算筛选后挑选了37个突变体进行实验验证,最终获得了高对映体选择性生产二元醇的柠檬烯环氧化物水解酶突变体。
在酶的热稳定性提升上,Janssen 等在多种天然脱卤酶的改造中运用其提出的FRESCO 策略[87],获得了很多成功案例[88-89]。
Arnold 等将SCHEMA 应用在beta-内酰胺酶[53]、人源精氨酸酶I和II[90]、真菌来源的木质纤维素酶[91]等的连续序列元素的重组改造中,均获得优异的突变体。
Li等[92]利用Rosetta设计软件对芽孢杆菌来源的天冬氨酸酶YM55-1 进行基于已有骨架的再设计,获得的再设计的酶能够催化碳-碳双键的不对称氢胺化,生产中酶的底物耐受浓度高达300 g/L,转化率、区域选择性、立体选择性均超99%。
以天然的酶骨架作为设计基础,结合前期的实验数据和理解总结,是计算机介入酶的设计的起点,越来越多地应用于实际酶的改造生产。
2.2 从头设计
从简单的αβ 折叠[93],到重复单元蛋白(repeat protein)[94]、自组装的纤维蛋白[78]、跨膜蛋白[95]、白细胞介素模拟物[96],再到最新的SARS-CoV-2 病毒蛋白抑制剂[76],Baker 及其团队在从头设计的领域一路披荆斩棘。蛋白序列空间示意如图3(a),Rosetta 从头设计方法如图3(b)。以一个200个氨基酸的天然蛋白为例,应该有20200个可能的氨基酸序列,而在自然进化过程中得到的天然蛋白只是对其中极小的一部分进行采样,定向进化方法扩大了序列采样空间,而蛋白的从头设计就是基于指导蛋白折叠的物理原理探索蛋白全序列的空间。科学家们希望利用从头设计的方法设计出自然界没有的蛋白,进而解决能源和医疗中人类共同面临的挑战。
2008 年,Baker 等基于从头设计的方法,成功设计出接近原子级精确度的非天然酶——Kemp 消除酶[85]和Retro-Aldol 酶[84],这是计算机科学和酶工程的一个重要里程碑。
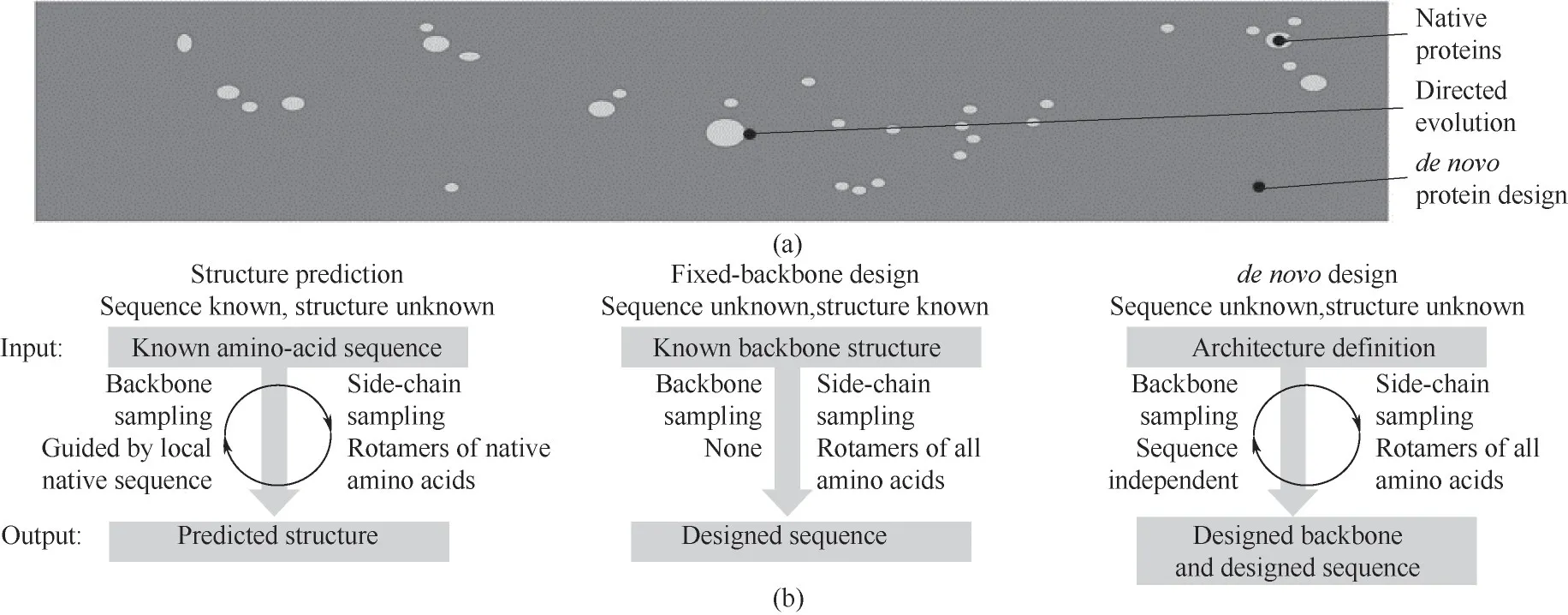
图3 蛋白的从头设计方法[97](a)蛋白序列空间示意图;(b)结构预测、固定骨架设计、从头设计方法Fig.3 Methods for de novo protein design[97](a)a schematic of the protein sequence space;(b)structure prediction,fixed-backbone design and de novo protein design methods
围绕Kemp 消除酶,Khersonsky 等[98-100]又展开了很多的研究来提升其催化效率。选择Glu、Trp、Ser和His-Asp、Phe、Ser 两种模式的催化位点,结合多种的骨架设计多轮突变,最终获得的最优突变体的kcat/Km值大于105L/(mol·s),较原始设计酶提升了5个数量级。Blomberg 等[101]利用Asp、Trp、Ser 三个氨基酸为原始活性位点,将Kemp 消除酶HG3 引入一个已知结构的耐高温的木聚糖酶(PDB:1GOR)骨架,通过骨架修饰和多次突变,获得的HG3.17 突变体使得底物(5-nitrobenzisoxazole)的质子传递速度提升了6×108倍,获得的晶体结构也与设计的结构几乎一致。
Diels-Alder 反应是一个一步完成的环加成有机反应,Siegel 等以Gln、Tyr 为活性位点,在二异丙基氟代磷酸酶(diisopropyl-fluorophosphatase)的骨架上设计出了具有高选择性和底物特异性的Diels Alderase[102],随后又用Fodit 进行骨架重构,酶活提升了18倍[103]。Świderek 等[42]对利用计算机设计的具有不同新功能的酶的发展、针对同一个反应不同的研究人员采用的不同算法和活性位点处理方式做了详细的介绍。
Lapidoth 等[104]将TIM-barrel 骨架按不同的方式进行拼接获得不同的骨架,再将活性位点引入,通过序列设计、应用可溶性提升算法,分别设计了43个木聚糖酶(glycoside hydrolase 10 xylanases,GH10)和34 个类磷酸三酯内脂酶(phosphotriesterase-like lactonases,PLL),最终获得21个GH10 和7个PLL设计具有催化活性,其中最优的四个设计的酶活性与其家族的天然酶相当。
金属离子在天然酶中扮演着重要的作用,自然界中有几乎半数的酶是结合了金属的蛋白,所以金属酶的从头设计也是从头设计研究的热点[105-106]。金属蛋白的主要特征就是作为一个金属配体复合体固定在宿主的生物分子上,固定的方式分共价键结合和非共价键结合,其中共价键结合往往是金属离子通过一个半胱氨酸残基成键[107]。
很多现代蛋白从原始序列印记追溯分析其起源似乎是简单的多肽。为了探究金属酶的进化过程,Studer 等[108]从一个具有酯键水解功能的多肽MID1出发,通过计算机设计,将MID1改造成能自组装成同源二聚体结构且拥有两个锌离子结合位点的多肽,又利用Gly-Ser-Gly 将二聚体亚基相邻的N和C 端连接起来,按照计算结果替换离连接肽较远端的锌离子结合位点,获得的变体MID1sc不仅能够结合一个二价锌离子,且拥有与初始多肽相似的催化乙酸对硝基苯酯水解的活性。
Bos 等[109]以环戊二烯与氮杂查尔酮类为底物的Diels–Alder反应为基础,在乳球菌多药耐药调节剂LmrR的二聚体界面上引入活性位点,使用半胱氨酸偶联策略,将铜(Ⅱ)菲咯啉复合物共价锚定在蛋白质的疏水性口袋中。合成的全新人工金属酶表现出高达97%的对映体选择性和非对映体选择性。
以人工金属蛋白为例,完全从头设计和基于已知蛋白的再设计的边界并不是特别地清晰[110]。此外,尽管已经在计算机设计酶上有了很多成功案例,但是酶的设计因其复杂性依然是非常具有挑战的工作。
3 人工智能酶设计
2020年12月,AlphaFold2在第14届国际蛋白质结构预测竞赛(CASP14)上向世界展示了人工智能在蛋白结构预测上可与核磁共振或X 射线晶体学、冷冻电子显微镜等实验技术相媲美的高正确率。机器学习(machine learning)是一种实现人工智能的方法,而深度学习(deep learning)是机器学习的子领域。与传统的以生物物理知识为基础的计算机设计方法不同,以深度学习[111]为例,在训练蛋白序列和结构时,不需要对蛋白的结构功能有深入的了解,便能寻找最好的折叠方式,预测蛋白的结构和功能,甚至是构建自然界未有的蛋白。
基于机器学习的酶设计方法流程如图4 所示[112],一般分为三个步骤。首先,数据准备与拆分,从实验中获得的大量数据被合理地统计于表中,之后将数据分为训练集(training set)与测试集(test set)两部分;其次,预测方法在训练集中进行模型训练;最后,利用测试集数据对模型进行测试与验证。
机器学习技术目前已经被应用于辅助预测酶结构设计[113],通过从蛋白酶数据中寻找特征模式(pattern model),以提高酶的稳定性、可溶性和活性,以及预测酶底物特异性[114]。
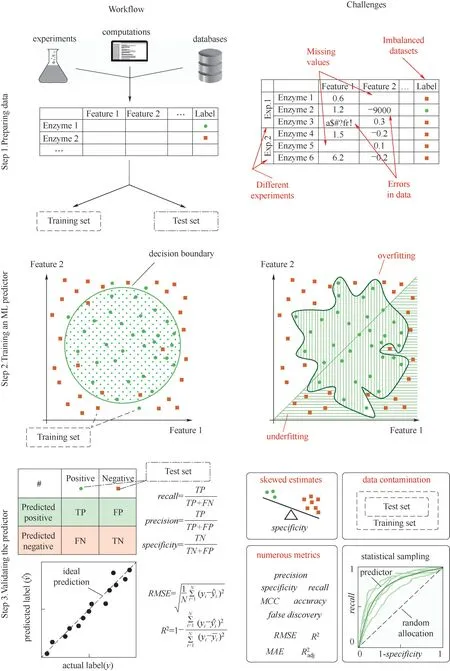
图4 构建机器学习预测器的工作流程和相关挑战[112]Fig.4 Schematic workflow of constructing an machine learning predictor and associated challenges [112]
UniRep(unified representation)是Alley 等[115]开发的一种深度学习的方法。计算机通过学习无标签的氨基酸序列,将提取蛋白的特征值变成一个富含有结构信息、进化信息和生物物理信息的统计表示(statistical representation)。Alley 等认为UniRep在预测自然界中或从头设计的蛋白的结构稳定性和定量突变体功能上接近最高水准。
Tallorin 等[116]基于机器学习方法开发了一种鉴定酶的特异性短肽(8~20 个氨基酸残基)底物的方法——POOL(peptide optimization with optimal learning)。POOL 利用迭代机器学习方法,在输入实验数据后,数学算法将会选择具有潜力的短肽底物作为接下来实验的对象,并且新的实验数据也会用于未来的算法优化过程。研究人员将该方法应用于鉴定两类4'-磷酸泛酰巯基乙胺基转移酶(4'-phosphopantetheinyl transferase, PPTase)的 短 肽 底物,这些短肽底物符合下列标准:可被PPTase 于保守的丝氨酸位点共价修饰来自CoA 的磷酸泛酰巯基乙胺;具备正交性,给定的短肽只能是某一类PPTase 的底物而非另一类的底物。通过POOL 方法,研究人员鉴定出了Sfp 类酶(来源于Bacillus subtilis的表面活性素磷酸泛酰巯基乙胺基转移酶)及AcpS 类酶(来源于Streptomyces coelicolor的全酰基载体蛋白合成酶)的短肽底物,并在纤维素膜阵列上利用形成图样(“A”或“S”)的方式证实了两类酶的短肽底物的良好正交性。
基于蛋白质序列及功能进行训练的机器学习方法可以在无物理或生物学等先验知识的情况下,推断未知蛋白质序列的生物学特性,也能够预测和发现具有功能的蛋白质。Yang 等[117]提出使用蛋白质嵌入序列(embedding sequence)作为机器学习的输入数据,使用蛋白质嵌入数据训练的机器学习模型,输入数据尺寸与直接使用序列相比减少几个数量级,但模型的预测能力和准确性可与现有模型相媲美。另外,他们还利用机器学习辅助定向进化来测试更大序列空间的突变,以降低实验成本[118]。
Yang 等[119]利用深度神经网络用序列比对的氨基酸残基共进化信息来推断距离约束,加上一个Rosetta-constrained 能量最少化程序快速生成模型从而预测蛋白结构,新的trROSETTA 模型能够实现从头设计蛋白的结构预测和突变体的影响捕捉。虽然该结构预测模型还没有涉及功能设计,但是结合传统的生物物理模型,将会有助于将新功能引入从头设计的蛋白。
机器学习流程中也存在着相应的困难如图4,如数据的缺失与错误,以及不同实验来源造成的数据不一致是模型训练中常见的问题;模型训练中的过度拟合(over fitting)与拟合不足(under fitting)也是基于机器学习的酶设计方法需要面对的挑战之一。目前并没有普适性的机器学习模型,模型的选择需要根据实际的设计目的而定,或者尝试多种算法挑选最优模型。
随着高通量测序与筛选等实验技术的发展与革新,相当庞大的高质量酶分子相关数据不断地积累。随着如深度学习等更复杂的机器学习方法的发展,基于机器学习的预测方法将会更加具有可靠性。可靠的机器学习方法为人工智能酶设计提供了起点,也为研究酶的结构-功能分子机理提供更多的机会,相信在不久的将来,将能够实现准确高效的人工智能的酶设计。
4 结 语
酶作为生物催化剂已经被广泛地应用在包括能源、医药、食品在内的许多领域。21 世纪以来,生物信息学和计算机科学的发展,为酶工程提供了全新的改造方法,扩大了酶的改造空间,优质酶的开发所需的时间和经济成本随着计算设计精度的提升而降低。可以预期未来,计算机硬件的提升和算法的优化,结合生物物理知识将会带来更精妙高效的酶活性位点的设计方法,解决人工设计的酶初始催化效率不高的瓶颈问题。将来甚至可以根据需求,快速设计精巧的新酶分子,应对人类面临的医疗、能源等领域的全新挑战。
猜你喜欢
分子催化(2022年1期)2022-11-02
中国音乐学(2022年1期)2022-05-05
昆明医科大学学报(2021年3期)2021-07-22
烟草科技(2021年6期)2021-06-24
教育周报·教育论坛(2020年3期)2020-10-21
科学(2020年2期)2020-08-24
电脑知识与技术(2018年19期)2018-11-01
科技资讯(2018年16期)2018-10-26
教学与管理(理论版)(2017年7期)2017-08-11
科技资讯(2017年12期)2017-06-09
