
热敏感相关癫痫的早期识别与诊断
2021-07-20 02:32:06徐瑜欣综述钟建民审校
中国当代儿科杂志 2021年7期
徐瑜欣 综述 钟建民 审校
(江西省儿童医院神经内科,江西南昌 330006)
[中国当代儿科杂志,2021,23(7):749-754]
热敏感相关癫痫是指一些癫痫以热性惊厥(febrile seizures,FS)起病,具有“热敏感”的特点,早期难以与FS鉴别,需要到一定年龄后表现出相应特征才能诊断。热敏感相关癫痫主要包括遗传性癫痫伴热性惊厥附加症(genetic epilepsy with febrile seizures plus,GEFS+)、Dravet综合征(Dravet syndrome,DS)、PCDH19基因相关癫痫等[1-2]。本文旨在总结热敏感相关癫痫的早期临床特点(表1),以期为临床医师早期识别热敏感相关癫痫提供一定依据,从而达到早期识别、早期诊断,有助于临床治疗,改善长期预后。
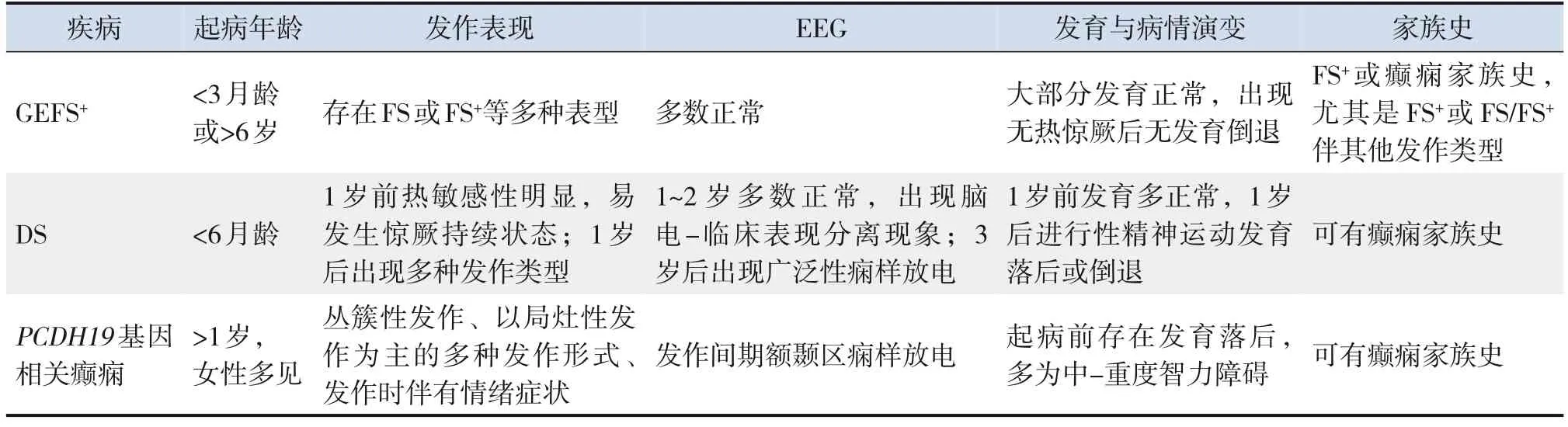
表1 GEFS+、DS、PCDH19基因相关癫痫的临床特征
1 GEFS+
GEFS+是一种以家族为整体进行诊断的癫痫综合征,1997年在一个澳大利亚家系中被首次报道[3],最初被称为全面性癫痫伴热性惊厥附加症(generalized epilepsy with febrileseizuresplus),随后的研究发现该疾病家系中可出现局灶性癫痫的患儿。因此,Scheffer等[4]建议将“全面性”(generalized)改为“遗传性”(genetic)。GEFS+在2001年被国际抗癫痫联盟定义为一种新的癫痫综合征[5],但目前尚无GEFS+发病率的流行病学调查,因为GEFS+通常在大家系中被诊断,而那些累及个体较少的家系常被漏诊。
既往研究认为GEFS+为常染色体显性遗传性疾病,外显率为62%~76%,但近年来的研究发现在较小的家系和散发病例中,GEFS+可呈多基因遗传或新生突变,还有两个近亲家系符合常染色体隐性遗传[6-7],因此其遗传方式极其复杂。目前分子遗传学研究认为,20%~30%的GEFS+家系能找到致病基因,仍有70%~80%的家系无法找到相应致病 基 因[6,8],GEFS+的 主 效 基 因 包 括SCN1A、SCN2A、SCN1B、SCN9A、GABRG2、STX1B,易感基因包括GABRD、CACNA1H和HCN2719_721 del PPP[7,9-10]。SCN1A是继SCN1B后第2个被发现与GEFS+相关的基因,也是最常见的致病基因,约19%的家系与SCN1A基因突变有关[6]。GEFS+家系中的SCN1A突变多为错义突变,且突变位于钠离子通道蛋白核心区(S4~S6)以外[7-8],王波等[11]研究发现SCN1A的rs3812718位点多态性与GEFS+易感性及FS/热性惊厥附加症(febrile seizuresplus,FS+)的临床表型相关。SCN1B是最早被证实与GEFS+相关的基因,约8%的家系存在SCN1B突变[6],该基因突变的FS/FS+患儿的FS起病高峰为24月龄,略晚于普通人群(18月龄)[7]。Myers等[12]研究表明SCN1B突变可能会增加GEFS+患儿的癫痫猝死风险。GABRG2基因在GEFS+家系中的突变率约9%[6],突变类型主要有无义、错义、剪切突变等,无义突变的患儿多为严重表型,而错义突变的患儿表型相对较轻。SCN2A为SCN1A的同源基因,其突变多与FS表型相关[10,13]。目前,国内外文献还显示SCN9A、STX1B等基因突变也与GEFS+有关,但在GEFS+家系中的筛查阳性率均<5%[14]。
GEFS+不仅存在基因异质性,而且在同一家系同一遗传基因中也存在较大的表型差异。FS是GEFS+表型谱中最常见的临床表型,占家系患儿成员的41%,起病中位年龄为12~24月龄[6]。第二常见的表型是FS+,占27%,是一种轻型的遗传性癫痫,存在以下几种不同表现形式:(1)最常见的是FS持续至6岁以后;(2)在3月龄至6岁期间既有FS又有无热全面强直-阵挛发作(afebrile generalized tonic-clonic seizures,AGTCS);(3)FS缓解数年后出现AGTCS,期间有无惊厥发作期;(4)FS起病年龄小于3月龄或大于6岁[6]。其他少见的表型还包括FS/FS+伴其他全面性发作,如FS/FS+伴失神发作、FS/FS+伴肌阵挛发作、FS/FS+伴失张力发作,共占5%[6]。不同于典型的失神发作,GEFS+家系中的失神患儿通常是1年发作数次,而不是每天发作多次[6,8]。除全面性癫痫外,约9%的GEFS+家系中可见局灶性癫痫患儿,如额叶癫痫和颞叶癫痫,其中包括伴海马硬化的内侧颞叶癫痫。还有3%的GEFS+家系中有仅表现为AGTCS的患儿,而无FS史,起病中位年龄为4岁[6]。GEFS+患儿的脑电图(electroencephalogram,EEG)改变主要取决于个体的临床表型,FS的EEG通常是正常,FS+可以是正常或广泛性不规则的棘慢波。头颅影像学检查通常是正常,但一些局灶性癫痫患儿也可出现海马硬化[15]。
GEFS+并非为个体诊断名称,是一个以家系为整体进行诊断的家族性癫痫综合征。其诊断标准一直在不断更新之中,最新GEFS+诊断标准分为两种情况:(1)家族中有FS+个体时,需要≥2位成员符合GEFS+表型谱,且其中至少有1位为FS+表型;(2)家族中没有FS+个体时,则需要≥3位成员符合GEFS+表型谱[6]。GEFS+患儿的治疗取决于其临床表型,对于仅有FS/FS+表型的患儿,应注重随访观察和家属的健康教育,必要时可予地西泮、左乙拉西坦、丙戊酸钠等抗癫痫药物治疗。FS/FS+伴其他全面性或局灶性癫痫的药物选择,则和其他癫痫一样根据发作类型选择治疗药物[15]。GEFS+家系中的患儿总体呈良性经过,智力运动发育正常,大多数在儿童后期或25岁前不再发作,但表型为DS或肌阵挛站立不能性癫痫的患儿,则预后不良[15]。
因此,对于有FS+或癫痫家族史的FS患儿,需要对病史和家族史进行详细询问和记录分析。当家系中有成员表现为FS+或FS/FS+伴其他发作类型时,应考虑GEFS+诊断的可能。
2 DS
DS既往又称婴儿严重肌阵挛癫痫是婴儿期起病的难治性癫痫性脑病,以难治性癫痫、认知障碍和运动障碍为主要特征。文献报道的发病率为1∶15 700~1∶40 900,无明显性别差异[16]。
目前已报道的DS相关致病基因包括SCN1A、GABRA1、STXBP1、CHD2、SCN1B、SCN2A等,其中SCN1A突变占80%[17],且90%为新发突变,10%为遗传性突变,SCN1A编码Ⅰ型钠通道(Nav1.1)α亚基,研究发现该基因的表达随着年龄增长而增加[17],故DS患儿常在婴儿期出现惊厥,并随着年龄增长癫痫发作增多。DS患儿的SCN1A突变多位于核心区域(S4~S6),以无义突变和移码突变多见,常常导致蛋白功能丧失[17]。田小娟等[18]研究发现携带SCN1A错义突变者、遗传性突变者发作控制相对较好。
DS患儿的临床表现可分为3个阶段[19-20]:(1)发作起始期:患儿在1岁以内常以FS起病,起病高峰年龄4~8个月,多具有长时间的全面性或半侧阵挛发作、强直-阵挛发作、1次热程中反复发作、易发生热性惊厥持续状态的特征,约94%的患儿至少有1次因长时间的癫痫发作而住院治疗[21-22]。热敏感性是DS患儿的重要特点,低热、接种疫苗、热水浴均可诱发发作[18]。患儿起病前语言、运动发育正常,但也有报道提出起病早期可出现一些轻微的异常,主要影响视觉和运动协调[23]。此期头颅磁共振成像检查正常,76%的患儿EEG无异常[24]。(2)恶化期:此期通常在1~5岁之间,患儿逐渐出现多种癫痫发作形式,且发作更加频繁,以肌阵挛发作、不典型失神发作、局灶性发作、全面强直-阵挛发作最常见,失张力发作少见[22]。>50%的患儿在此阶段出现癫痫持续状态(status epilepticus,SE),多在6~18月龄[21]。DS患儿在1岁以后逐渐出现进行性精神运动发育落后或倒退,尤其是语言发育迟缓,可伴运动障碍、孤独症谱系障碍、行为异常和步态异常[24]。同时,发作间期EEG的背景活动逐渐恶化,清醒期出现双侧中央、顶区为主的5~7 Hz的θ节律长程发放。虽然1~2岁间发作频繁且难以控制,但仅有50%的患儿出现痫样放电,这种早期脑电-临床表现分离现象是考虑DS的线索之一[24]。3岁以后,90%以上的患儿会出现EEG背景异常或痫样放电,以广泛性棘慢复合波、多棘慢复合波为主,也可出现局灶性或多灶性放电[24-25]。(3)稳定期:该阶段的早期(一般<10岁),癫痫发作的频率和严重程度逐渐降低,患儿的热敏感性仍然存在,但灵敏度降低,最常见的发作类型为全面强直-阵挛发作,且主要发生在睡眠期[22,26],肌阵挛发作、非典型失神发作和伴意识改变的局灶性发作较少见。随着其他癫痫共患病的恶化(如发育迟缓、异常步态和共济失调),认知障碍和语言障碍变得越来越明显[19,27]。
DS是一种临床诊断性疾病,多在2~4岁出现典型临床表现时才被诊断。然而,早期诊断和治疗,可能改善长期预后,减轻认知、运动和行为损伤的严重程度。Cetica等[28]发现SCN1A突变患儿中,起病年龄为0~6月龄者发展成DS的风险为85%,6~12月龄者占51%,>12月龄则为0%,且起病年龄6月龄作为DS预测指标的灵敏度为83.3%,特异度为76.6%。该研究显示相比剪切位点突变和错义突变,SCN1A发生截短突变的患儿更易发展为DS(40%vs 13%)[28],但截短突变作为DS预测指标的灵敏度为80.85%,特异度仅有48.61%。Xu等[29]总结了138例DS患儿的临床特征,并提出在1岁前出现发热诱发发作的患儿中,具备≥2条复杂性FS特点者,应考虑DS诊断的可能。Hattori等[30]制定了一个DS风险评分筛查表:FS起病年龄≤7个月(2分);1岁内惊厥总次数≥5次(3分);偏侧惊厥(3分);局灶性发作(1分);肌阵挛发作(1分);热水浴诱发发作(2分),总分≥6分是预测DS的高危临界值,其灵敏度和特异度分别为98%、94%。北美共识小组[16]推荐当患儿在1岁以内出现≥2次长程发作(>15 min)的FS,其中至少有1次表现为局灶性发作,而头颅磁共振检查正常,发育正常时,需警惕DS的可能,并建议进行SCN1A突变筛查。de Lange等[31]研究提示首次无热惊厥的年龄是预测DS的一个可靠指标,首次无热惊厥发生在24月龄前的SCN1A突变患儿,约96%最终发展为DS,而24月龄后发生首次无热惊厥的患儿仅有20%发展为DS。
所以,具有起病年龄在6月龄以内、1岁前发生≥2次长程发作的FS(>15 min)、表现为局灶性发作或SE的FS患儿,或24月龄前出现无热惊厥的FS患儿,需警惕DS可能,并完善基因筛查进行早期预测,对此类患儿进行密切随访观察,出现相应临床表现时及时干预,以改善长期预后。
3 PCDH19基因相关癫痫
PCDH19基因位于Xq22.1,编码的蛋白介导同型细胞间的结合,其主要在中枢神经系统表达[32-33]。PCDH19突变大多数发生在第1外显子,突变类型包括错义突变、无义突变、碱基缺失或插入突变、剪切位点突变,部分外显子甚至整个基因的缺失或重复,其中50%以上为错义突变[32,34]。虽然PCDH19突变2008年首次被Dibbens等[35]以限于女性的癫痫伴智力低下的家系报道,但约50%的患儿为新发突变[36-37]。PCDH19基因相关癫痫具有特殊的X连锁遗传方式,表现为女性杂合子或男性嵌合体发病,而男性半合子不发病。目前有多种假说,最被广泛认可的是“细胞干扰”假说,即只有PCDH19基因突变型和野生型两种不同的细胞共存时,患儿才会发病[32,34,36]。
PCDH19基因相关癫痫的临床表型具有异质性,同卵双胞胎可出现不同表型。核心特征包括早发性癫痫发作、热敏感性、丛簇性发作,起病年龄在1~70月龄,高峰年龄为10~12月龄[34,38-39]。多数PCDH19基因相关癫痫患儿具有2种及2种以上的发作类型,63%表现为局灶性发作,全面强直-阵挛发作、强直发作、阵挛发作、不典型失神、肌阵挛发作等其他发作类型相对较少,失张力发作更为少见,约71%的患儿发作时伴有惊恐尖叫等情绪症状[34,38,40]。50%的患儿以FS起病,且90%左右发热时会诱发或加重癫痫发作,也有研究表明部分患儿以无热惊厥起病,2岁以后出现典型的热敏感性,此后热敏感性随着年龄增长而降低[38,41]。丛簇性发作是PCDH19基因相关癫痫的显著特征,可出现于46%~92%的患儿,单次发作持续时间较短,大部分小于1 min,出现的频率从每天到每年不等,50%的患儿每1~3个月出现一次丛簇性发作,持续数小时或数天[38,42]。30%的患儿表现为SE,其中37%为非惊厥性癫痫持续状态,63%为惊厥性癫痫持续状态,表现为频繁反复发作性短时间惊厥,但发作间期意识不能恢复[42]。大部分患儿10岁以后癫痫发作的频率逐渐降低,可能与青春期开始前的激素水平改变有关[43],但认知障碍和行为障碍成为主要的临床问题。47%~75%的PCDH19基因相关癫痫患儿会有中-重度智力障碍,仅少数可无智力障碍[42]。此外,55%左右会出现行为障碍,主要表现为刻板行为、强迫行为、攻击性行为等,但目前发现认知功能与癫痫的严重程度无明显相关性[42]。
PCDH19基因相关癫痫患儿的EEG尚未发现有年龄依赖性。约82.8%局灶性发作起源于颞区,在同一次丛簇性发作中,可起源于不同的半球,10%左右的患儿在闪光诱发试验中出现光阵发性反应[42]。51%~64%的患儿发作间期EEG可出现痫样放电,以额颞区为主,可能与PCDH19在海马形成相关的区域中显著表达有关[44]。
Higurashi等[45]报道在急性丛簇性发作时,静脉使用皮质类固醇激素能迅速缓解症状,但在数周内会再次出现癫痫发作。研究发现最有效的药物为氯巴占和溴化钾,1年后的有效率分别为50%和43%[34]。
综上所述,1岁后起病,发作具有热敏感性和丛簇性、抗癫痫药物效果不佳的女性患儿可做为PCDH19基因相关癫痫的重要线索,基因检测提示PCDH19突变可进行早期诊断。
4 总结与展望
热敏感相关癫痫在起病早期不易与FS区分,容易被误诊、漏诊。本文总结了GEFS+、DS、PCDH19基因相关癫痫的临床特征,以期为临床医师早期识别热敏感相关癫痫提供一定依据,必要时早期进行基因检测辅助诊断,做到早诊断、早治疗,且避免使用加重癫痫发作的药物,减少不必要的医疗花费,改善患儿的长期预后。同时,尽早向家属解释患儿病情演变规律、家庭护理及预后同样具有重要意义。
猜你喜欢
中国临床医学影像杂志(2021年10期)2021-11-22 07:46:44
中国临床医学影像杂志(2021年6期)2021-08-14 02:21:50
科学生活(2019年7期)2020-01-01 08:28:02
现代临床医学(2019年4期)2019-09-10 07:43:46
养殖与饲料(2019年10期)2019-02-25 14:52:37
山东畜牧兽医(2018年3期)2018-04-26 09:10:34
广东海洋大学学报(2015年4期)2016-01-13 08:39:30
听力学及言语疾病杂志(2015年5期)2015-12-24 01:47:04
听力学及言语疾病杂志(2015年5期)2015-12-24 01:47:04
首都医科大学学报(2015年4期)2015-12-16 13:00:08
