
儿童EB病毒相关平滑肌肿瘤1例
2021-07-20 02:32:06师琴汤文芳贺湘玲田鑫
中国当代儿科杂志 2021年7期
师琴 汤文芳 贺湘玲 田鑫
(湖南省人民医院/湖南师范大学附属第一医院儿童医学中心血液肿瘤科,湖南长沙 410005)
[中国当代儿科杂志,2021,23(7):739-742]
患儿,女,6岁11月龄,因双下肢疼痛1年余入院。1年前患儿无明显诱因出现双下肢疼痛,以髋关节、膝关节为主,阵发性发作,每次持续时间约1~2 min,卧床休息稍缓解,晨起疼痛明显,偶诉头痛,无发热、咳嗽,无呕吐、抽搐等不适。患儿病情反复,外院治疗效果不佳,遂至我院就诊。既往有血小板减少病史,外婆有“关节炎”病史,个人史无异常。入院体检:T 36.7℃,P 96次/min,R 22次/min,BP 98/60 mm Hg,体重16 kg。神志清楚,颈部及腹股沟处可扪及肿大淋巴结,最大者约1.0 cm×1.0 cm,质软,无压痛,皮肤无红肿,活动度可。头部、口咽、心、肺、腹体检无异常。脊柱生理弯曲正常,四肢肌力、肌张力正常,无活动受限。辅助检查:EB病毒抗体:EB-VCA-IgG>750 U/mL(参考值:0~20 U/mL),EB病毒DNA正常,乙肝病毒DNA 3.79×107IU/mL(参考值:检测下限10 IU/mL);淋巴细胞亚群检测:辅助/诱导T淋巴细胞百分比23.97%(参考值:31.7%~47%)、总T淋巴细胞百分比49.6%(参考值:63.2%~77.8%)、辅助/诱导T淋巴细胞绝对数目516个/μL(参考值:646~1 515个/μL)、总T淋巴细胞绝对数目1 030个/μL(参考值:1 239~2 611个/μL);艾滋病毒抗体、梅毒螺旋体抗体、丙肝抗体阴性,血常规、凝血功能、风湿狼疮全套、免疫功能、细胞因子、血生化、中性粒细胞呼吸爆发试验、红细胞沉降率均正常。胸部CT平扫示右肺下叶外基底段及左肺上叶前段高密度结节灶,直径分别为7 mm、4 mm(图1A~B)。腰椎MRI示L2椎体变扁,以左侧稍明显,椎旁(L2椎体左侧及后方)团块状软组织肿块,伴L2椎体左侧附件受累,椎管稍狭窄改变(图1C)。头颅MRI T2加权像示双侧海绵窦区结节状短T2信号灶,右侧较大,直径为17 mm(图1D)。腰椎病变组织病理:EB病毒相关平滑肌肿瘤,瘤细胞轻度异型,未见明确核分裂,Ki67增殖指数低,未见明确肿瘤性坏死,可见骨质破坏,形态学属于高分化。免疫组化:Ki67散在阳性,SMA弥漫阳性,CD34(血管+),Desmin(-),CD117、S-100蛋白、DOG1、ALK均阴性;原位杂交示EB病毒编码的RNA阳性。通过靶向捕获基因测序发现患儿ITK基因存在c.725_730delAGAGTA(p.K242_S243del)缺失突变,目前未见报道,该位点6个碱基缺失(AGAGTA),导致蛋白缺失第242位的赖氨酸和第243位的丝氨酸;经家系Sanger验证分析,患儿父母该位点为杂合变异(图2)。根据美国医学遗传学与基因组学学会发布的遗传变异分类标准与指南[1],该变异致病性判定为临床意义未明(PM2+PM4)。
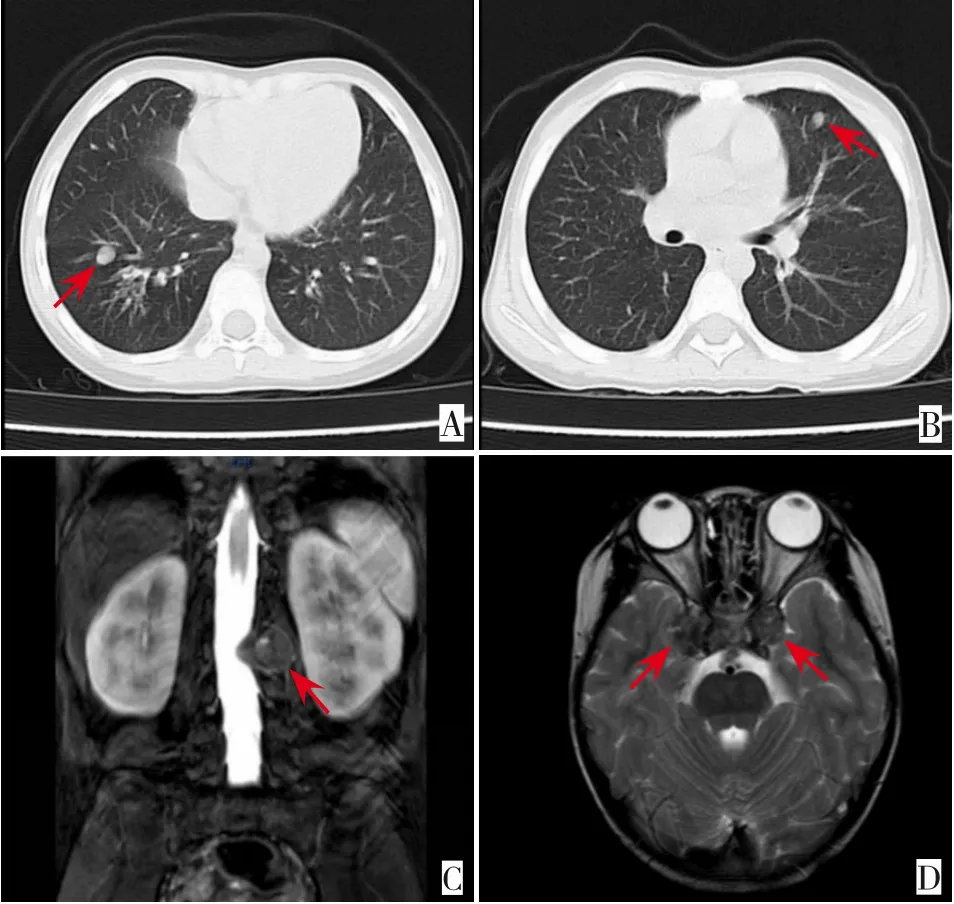
图1 患儿影像学结果 A为肺部CT肺窗,可见右肺下叶结节,直径约7 mm;B为肺部CT肺窗,可见左肺上叶结节,直径约4 mm;C为腰椎MRI,L2椎旁(L2椎体左侧)团块状软组织肿块,增强扫描呈明显强化;D为头颅MRI双侧海绵窦旁见结节状软组织肿块,右侧较大约17 mm。各箭头所指为相应病变位置。
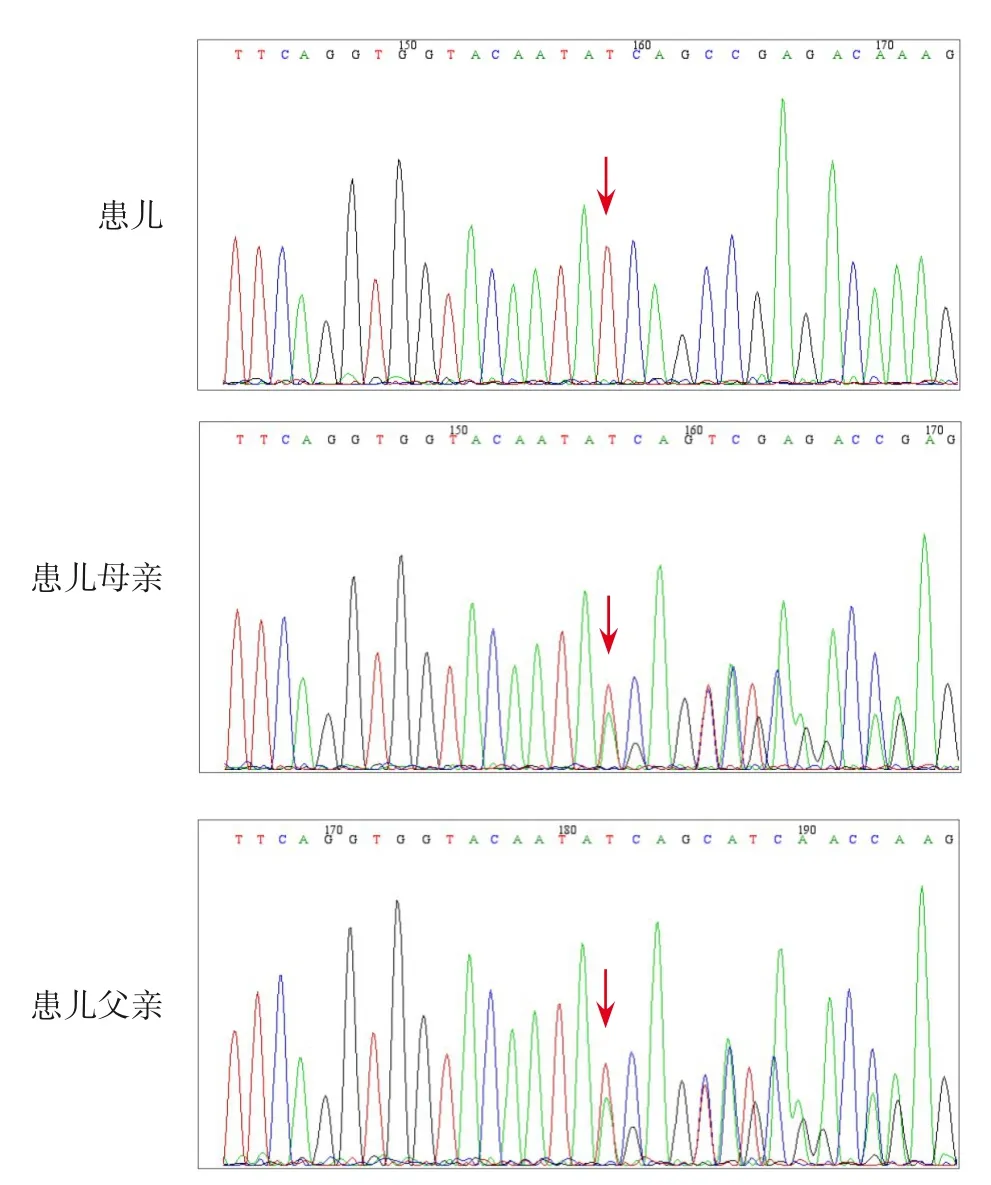
图2 患儿及其父母ITK基因Sanger测序图 患儿存在ITK基因c.725_730delAGAGTA纯合变异,其父母该位点均为杂合变异。突变位点如箭头所示。
患儿以双下肢疼痛为主要表现,考虑与腰椎区肿块压迫神经有关,请相关外科医师会诊后综合考虑因患儿头颅及肺部病灶小,可暂观察随访,腰椎区肿块较大,且有肿瘤压迫症状,予手术切除,术后患儿双髋关节疼痛基本消失。患儿出院后随访至今9个月,未诉双髋关节疼痛,头颅、肺部结节未见增大,未见复发及新发肿块。
讨论:EB病毒相关平滑肌肿瘤(Epstein-Barr virusassociated smooth-muscle tumor,EBV-SMT)是一种极罕见的肿瘤,在20世纪90年代被首次报道[2]。该病多见免疫缺陷的病例,如先天性免疫缺陷、获得性免疫缺陷综合征及移植后免疫抑制[3]。艾滋病患者发生EBV-SMT更倾向于中枢神经系统受累[4],移植后EBV-SMT常见发病部位为肝脏,先天性免疫缺陷EBV-SMT国内外少见报道,均发生于儿童,中位发病年龄为8岁,肺是最常见的发病部位[5]。
EBV-SMT临床表现主要取决于肿瘤的发生部位、肿瘤大小及相应器官移位及破坏程度[6]。肿瘤可发生在单个或多个器官,多部位的EBV-SMT病灶均为独立生长[7]。EBV-SMT影像学表现无特异性,不能靠影像学检查确诊。本例患儿以髋、膝关节疼痛为主要表现,病程1年余,腰椎、头颅、肺均可见肿块,腰椎区为甚,下肢影像学正常,结合临床表现及辅助检查,患儿双下肢间歇性疼痛考虑腰椎肿块压迫神经所致。基因检测显示该患儿存在ITK基因缺失突变,该基因编码在T细胞中表达的细胞内酪氨酸激酶,蛋白包含sh2和sh3两个结构域,通常存在于细胞内激酶中,在T细胞增殖和分化中起作用。本例患儿存在免疫缺陷相关基因ITK突变,该变异尚未被报道,临床意义虽尚未明确,但研究表明,ITK基因突变与T细胞原发性免疫缺陷和EB病毒-淋巴增生性疾病相关,ⅠTK功能的丧失可能导致T细胞对致癌病毒的反应失调[8]。患儿EB病毒抗体升高,DNA定量检测正常,提示存在EB病毒既往感染,处于免疫抑制状态,这可能与患儿肿瘤发生有关。但EB病毒感染与免疫缺陷患儿肿瘤的发生时间关系仍不明确[2],需进一步研究。
EBV-SMT常表现为轻至中度多形性非典型梭形细胞交织成束,细胞胞浆呈嗜酸性[7,9]。约一半病例可见由原始圆形细胞构成的结节,病灶内T细胞的出现也是一个常见的特征[6]。EBV-SMT主要靠免疫组化及原位杂交诊断。免疫组化可见到所有肿瘤细胞均表达肌动蛋白,结蛋白多数阳性,但也有文献报道结蛋白阴性病例,原位杂交显示EB病毒编码的RNA阳性[10]。研究表明,85%的艾滋病相关EBV-SMT、98%的移植后相关EBV-SMT及100%的先天性免疫缺陷综合征相关EBV-SMT的免疫组化结果显示EB病毒编码的RNA[11]。本例患儿腰椎病变组织病理结果符合EBV-SMT免疫组化及原位杂交表现,诊断明确。该患儿头颅及肺部肿块相对较小,且无相关临床症状,未行病理活检,但结合EBV-SMT的临床特点及患儿的临床表现,头及肺部肿块临床考虑为EBV-SMT可能性大,目前患儿定期行影像学检查随访,若肿块进展,必要时行病理活检进一步证实。
目前EBV-SMT的治疗尚无统一的治疗方案,现有文献报道的治疗方法包括手术切除治疗、减少免疫抑制剂的应用、抗病毒治疗。放化疗对病人一般无效,减少免疫抑制剂的使用可以恢复T细胞功能及诱导免疫反应,但对于移植后的病人可增加移植物排斥反应,导致移植失败[3]。手术切除仍是一线治疗,对于适合手术切除的病灶可尽早手术,如果肿瘤有症状且有颅内侵犯,全切除可提高生存率[4,12]。EBV-SMT患者AKT/mTOR通路被激活,抑制该通路可能具有治疗作用,西罗莫司是一种哺乳动物雷帕霉素靶蛋白抑制剂,有文献报道肿瘤在使用西罗莫司后自发消退[13],但其有效性仍需进一步研究。本例患儿手术完整切除腰椎肿块后肿瘤压迫症状消失,头颅、肺部的病灶小,且无相关肿瘤浸润症状,未行手术切除,目前随访无新发病灶及复发,头颅、肺部病灶无增大,一般情况良好。
综上所述,EBV-SMT是一种罕见的肿瘤,多发生于免疫功能缺陷患者,其发病机制目前仍存在争议,但免疫缺陷是患者发病的必要条件,特别是T细胞和NK细胞免疫对该肿瘤发病至关重要[14]。肿瘤的预后与患者基础疾病、器官累及程度有关。治疗以手术尽量完全切除肿块为主[4]。对于不明原因反复下肢疼痛,下肢影像学正常,且常规治疗无效的患儿,可尽早完善全身影像学检查及基因检测,以帮助早期诊断。
猜你喜欢
中华实用诊断与治疗杂志(2022年2期)2022-09-02 01:47:28
中老年保健(2021年6期)2021-08-24 06:55:22
浙江医学(2020年9期)2020-07-01 10:17:42
浙江中西医结合杂志(2019年4期)2019-05-05 10:51:52
浙江医学(2019年2期)2019-01-23 06:38:24
中国生殖健康(2019年11期)2019-01-07 01:27:38
中国卫生标准管理(2015年15期)2016-01-15 02:58:43
中国继续医学教育(2015年1期)2016-01-06 01:36:10
肝胆胰外科杂志(2015年4期)2015-02-27 11:12:29
中国中医药现代远程教育(2014年11期)2014-08-08 13:23:44
