
超支化聚酯阻垢剂的合成
2021-07-18 01:32王亚杰秦冬玲
南京工业大学学报(自然科学版) 2021年4期
王亚杰,秦冬玲,杨 刚
(南京工业大学 化工学院 材料化学工程国家重点实验室,江苏 南京 211800)
生产、生活中水被应用在各个环节,如冷却水循环、石油开采、污水处理、膜应用等过程。但水中含有的难溶性矿物质盐离子,会随着水体条件的改变形成水垢[1]。水垢会阻碍设备与水体间的能量传递过程,增加能耗,降低效率[2];会使管线输送不畅甚至堵塞;会黏附在设备表面造成腐蚀,严重时甚至发生重大安全事故[3-5]。因此,预防水垢是工业生产中必须解决的问题[6]。向水体中投加阻垢剂的预防方法因具有操作简便、不损伤管路和设备、经济有效等优点,得到广泛的应用[7-10]。
超支化聚合物具有由中心向空间外立体发散、类球状的高支化度的结构,内部含有大量的短支链和微小空腔,这一特点使其具有优良的吸附能力[11-12]。超支化聚合物外围有大量的活性端基官能团,可对其进行修饰,使超支化聚合物具有优异的阻垢性能。钱凯等[13]以丙烯酸甲酯和二乙烯三胺为原料合成了超支化聚酰胺,并对其端基进行羧酸化改性得到阻垢剂,当阻垢剂质量浓度为6 mg/L时,对CaSO4的阻垢率达到95 %以上,表明羧基具有高效螯合金属离子和分散微晶的能力。Huang等[14]通过迈克尔加成和酰胺化反应合成一系列具有超支化结构的阻垢剂(HBP),在pH为6~8的条件下,使用20 mg/L的HBP对CaSO4的阻垢率达到93.9 %。Zhang等[15]在140 ℃下、反应6 h合成了超支化聚酯,然后以三乙胺为催化剂,在105~110 ℃下改性6 h,得到端羧基超支化阻垢剂。当阻垢剂质量浓度为200 mg/L时,对CaCO3的阻垢率达到70%,具有一定的阻垢效果,但该阻垢剂合成和改性耗时较长,阻垢剂投加量较大且阻垢效果并不理想。
提高超支化阻垢剂的阻垢率,达到“低量高效”的效果,重在提高阻垢基团的含量。为此,本文通过一锅熔融缩聚法合成了端羟基超支化聚酯(HBPE—OH),首次用高性能催化剂4-二甲氨基吡啶(DMAP)对其端基进行羧酸化修饰,合成了HBPE—COONa阻垢剂。
1 实验
1.1 主要原料
2,2-二羟甲基丙酸(DMPA),化学纯,阿拉丁试剂有限公司;三羟甲基丙烷(TMP),化学纯,阿拉丁试剂有限公司;对甲苯磺酸(p-TSA),化学纯,安耐吉化学有限公司;4-二甲氨基吡啶(DMAP),化学纯,阿拉丁试剂有限公司;丁二酸酐(SA),化学纯,国药集团化学试剂有限公司;丙酮,分析纯,上海凌峰化学试剂有限公司;乙醚,分析纯,上海凌峰化学试剂有限公司;吡啶,分析纯,国药集团化学试剂有限公司。
1.2 超支化聚酯阻垢剂的合成
1.2.1 缩聚反应合成HBPE—OH
磁力搅拌条件下,在三口烧瓶中加入TMP(1.369 1 g,0.01 mol),DMPA(12.318 1 g,0.12 mol)和一定量的催化剂p-TSA,通入N2,在油浴锅中缓慢加热至一定温度使原料熔融。反应一定时间后切断N2,改成负压状态继续反应一段时间。反应结束后,将三口烧瓶从油浴锅中移出,冷却至室温,粗产物为淡黄色透明凝胶。向三口烧瓶中加入40 mL丙酮(无水),并转移至50 ℃水浴,产物溶解后,室温下缓慢倒入装有冰乙醚的烧杯中,沉淀并抽滤,得到白色固体,重复纯化3次,将产物在50 ℃真空干燥箱中干燥2 d,制得HBPE—OH。反应方程式如图1(a)所示。
1.2.2 羧酸化反应合成HBPE—COONa
称取HBPE—OH1.179 g(1 mmol)投入装有磁力转子的三口烧瓶中,加入SA 1.3 g(13 mmol,过量)和催化剂DMAP 0.07 g(n(—OH)的5%),在一定温度下回流反应一段时间。反应结束后冷却至室温,在冰浴条件下,将其缓慢倒入装有NaOH/无水乙醇溶液(0.1 mol/L)的烧杯中生成沉淀,抽滤并重复纯化3次,真空干燥箱中50 ℃下干燥24 h,得到绿色HBPE—COONa阻垢剂。反应方程式如图1(b)所示。

图1 阻垢剂合成反应式Fig.1 Synthesis reaction of scale inhibitor
1.3 阻垢剂的表征
将阻垢剂在60 ℃真空干燥箱中干燥12 h后,将其与KBr按质量比为1∶150的比例混合研磨并压成透明薄片,用美国Nicolet公司8700型号的FT-IR进行表征,通过出峰位置判断官能团;以氘代二甲基亚砜(DMSO)为溶剂,用德国Bruker公司AVANCEⅢHD 400M型号的核磁共振波谱仪(NMR)进行氢谱表征,通过峰型判断聚合物的结构;以重水为溶剂,用德国Bruker公司生产的AVANCEⅢHD 400M核磁共振波谱仪(NMR)进行碳谱表征,进而判断其支化度。
1.4 性能评价
1.4.1 羟值滴定
称取0.5 g左右HBPE—OH置于磨口锥形瓶中,加入25 mL乙酸酐/吡啶溶液(体积比1∶23),立即装上冷凝管并用吡啶封住接口,在油浴中缓慢升温到120 ℃并加热1 h,期间摇晃锥形瓶3次。然后从冷凝管顶部滴加10 mL去离子水并继续加热10 min,结束后取出锥形瓶放至室温,滴加5滴酚酞试剂并摇匀,用标定好的约0.5 mol/L的KOH/无水乙醇溶液进行滴定。做平行实验控制误差,并做空白实验。羟值计算公式见式(1)。
w(—OH)=(V0-V1)c×56.10/m
(1)
式中:w(—OH)为试样羟值,mg/g;V0为空白实验滴定所用的KOH/无水乙醇溶液的体积,mL;V1为样品实验滴定所用的KOH/无水乙醇溶液的体积,mL;c为标定后的KOH/无水乙醇溶液的浓度,mol/L;m为称取的试样质量,g。
1.4.2 静态阻垢实验
称取7.50 g NaCl、8.382 g无水CaCl2定溶于1 L容量瓶中配制成A溶液;称取7.50 g NaCl、10.66 g Na2SO4定容于1 L容量瓶中配制成B溶液;称取0.03 g阻垢剂定容于100 mL容量瓶中配制成C溶液。
取250 mL锥形瓶编号为1,向其中准确加入A溶液50 mL和C溶液0.5 mL,然后放在70 ℃烘箱中预热30 min。另取一250 mL锥形瓶编号为2,向其中准确加入B溶液50 mL和C溶液0.5 mL,放在70 ℃烘箱中预热30 min。取出两锥形瓶放至室温,将2号锥形瓶中溶液转移到1号锥形瓶中并称质量,结果精确到0.1 g。将1号锥形瓶放到70 ℃水浴中恒温24 h,然后取出放至室温,称质量,使锥形瓶前后质量一致。取水并用0.45 μm水系滤膜过滤后,用电感耦合等离子光谱仪(ICP)检测水样中的Ca2+质量浓度,另做空白实验。阻垢率公式见式(2)[16-17]。
η=(ρ2-ρ1)/(ρ0-ρ1)
(2)
式中:η为阻垢率;ρ0为空白实验恒温前的Ca2+质量浓度,mg/L;ρ1为空白实验恒温后的Ca2+质量浓度,mg/L;ρ2为加入阻垢剂且恒温后的Ca2+质量浓度,mg/L。
1.4.3 CaSO4垢样的制备

2 结果与讨论
2.1 反应条件优化
2.1.1 缩聚反应时间对阻垢效果的影响
在固定缩聚反应温度140 ℃、催化剂用量为反应单体(DMPA)质量的1%、固定羧酸化反应时间3 h、温度80 ℃、催化剂用量为n(反应底物)的5%(用量较少,不做考察)的情况下,考察缩聚反应时间对羟值和阻垢率的影响,结果见图2。
由图2可知:随着缩聚反应时间的延长,羟值和阻垢率都呈先增大后减小的相同趋势,在4 h时,羟值达到最大值550.8 mg/g,阻垢率达到最大值94.8%。这是因为反应时间短,反应单体没有足够的时间与主体反应,超支化结构向外扩散生长的支化结构较少,内部空腔较少,吸附和分散微晶能力较弱,因此阻垢率低;而反应时间过长,容易发生分子间和分子内成环反应[18],羟基被大量消耗而导致羟值降低,羟值的大小与羧基化后羧基含量有直接的关系,羟值小使得阻垢剂上的阻垢基团数量少,影响了阻垢效果。因此,最佳缩聚时间为4 h。

图2 缩聚反应时间对羟值和阻垢效果的影响Fig.2 Effects of polycondensation reaction time on hydroxyl value and scale inhibition effect
2.1.2 缩聚反应温度对阻垢效果的影响
在固定缩聚反应时间4 h、催化剂质量分数1%、固定羧酸化反应时间3 h、温度80 ℃、催化剂用量为n(反应底物)的5%的情况下,考察缩聚反应温度对羟值和阻垢率的影响,结果如图3所示。
本实验选取130~150 ℃为缩聚温度考察区间,因为低于130 ℃反应原料达不到熔融温度,反应无法进行;而温度过高则容易醚化。由图3可知:羟值和阻垢率均随温度的升高呈现先增大后减小的趋势,在140 ℃时,羟值达到最大值550.8 mg/g,阻垢率达到最大值93.7%。这是因为随着温度的升高,缩聚反应速率加快,同时副反应(醚化反应、酯交换反应、成环反应等)反应速率加快[19],导致副反应生成的小分子物质会被N2流或负压带出反应体系,因此反应结束后发现,在实验所用的蒸馏头和冷凝管内表面出现一层“白霜”。所以,缩聚反应温度选择140 ℃。
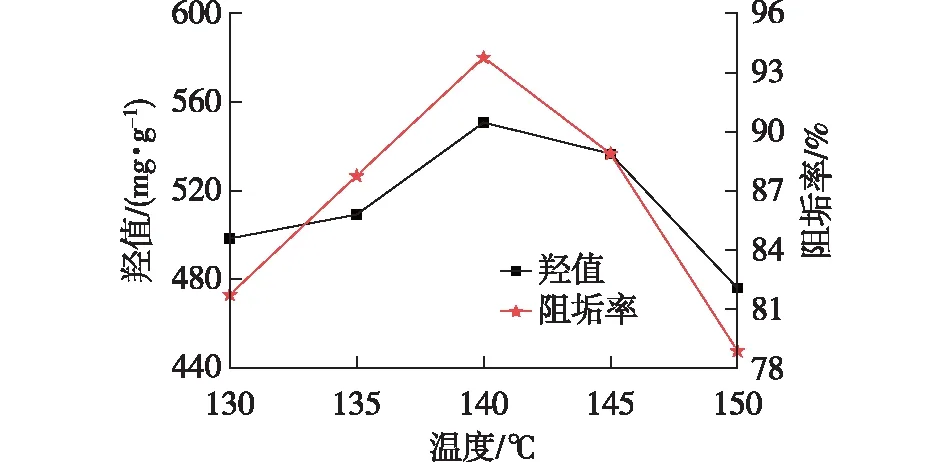
图3 缩聚反应温度对羟值和阻垢效果的影响Fig.3 Effects of polycondensation reaction temperature on hydroxyl value and scale inhibition effect
2.1.3 缩聚反应催化剂用量对阻垢效果的影响
在固定缩聚反应时间4 h、温度140 ℃、固定羧酸化反应时间3 h、温度80 ℃、催化剂用量为n(反应底物)的5%的情况下,考察缩聚反应催化剂用量对羟值和阻垢率的影响,结果如图4所示。
由图4可以看出,羟值和阻垢率均随催化剂量的增加先增加后减小,这是因为催化剂量小缩聚反应缓慢,在设定的反应时间内一些小分子聚合物来不及聚合到主体聚合物中,反应不完全,阻垢效果不理想;而催化剂量过大则副反应的速率亦会加快,且会导致HBPE—OH发生水解,影响产物收率。在催化剂质量分数为0.8%时,阻垢率最大值为96.4%,羟值却还在增大,在催化剂质量分数为1%时,达到最大值。这是因为阻垢效果不仅和端基含量有关系,还和分子结构(短支链和空腔)有关,短支链和空腔的增多有助于提高阻垢剂的吸附能力,能够容纳和分散更多的晶核,更好地抑制水垢的生成。因此,缩聚反应催化剂用量为DMPA质量的0.8%是最佳用量。

图4 缩聚反应催化剂质量分数对羟值和阻垢效果的影响Fig.4 Effects of mass fraction of polycondensation catalyst on the hydroxyl value and scale inhibition effect
2.1.4 羧酸化反应温度对阻垢效果的影响
在固定缩聚反应时间4 h、温度140 ℃、催化剂质量分数为1%、固定羧酸化反应时间3 h、催化剂用量为n(反应底物)的5%的情况下,考察羧酸化反应温度对羧酸化程度和阻垢率的影响,结果如图5所示。
由图5可知,羧酸化程度和阻垢率随反应温度的升高先急剧增加,在70 ℃达到最大值。随着温度继续升高,羧酸化程度和阻垢率开始下降,这是因为反应温度过高,会发生分子间酯化反应和分子内酯化反应,导致羧基减少,且使分子量急剧增加甚至凝胶化,反应结束后会发现在三口烧瓶底部有微黄色透明黏性凝胶颗粒的产生。而分子量过大时,端羧基会因为强极性作用而产生絮凝,降低了阻垢效果。因此,最佳羧酸化反应温度为70 ℃。
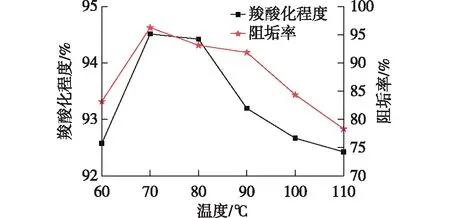
图5 羧酸化反应温度对羧酸化程度和阻垢率的影响Fig.5 Effects of carboxylation reaction temperature on carboxylation and scale inhibition rate
2.1.5 羧酸化反应时间对阻垢效果的影响
在固定缩聚反应时间4 h、温度140 ℃、催化剂质量分数为1%、固定羧酸化反应温度80 ℃、催化剂用量为n(反应底物)的5%的情况下,考察羧酸化反应时间对羧酸化程度和阻垢率的影响,结果如图6所示。
由图6可知,随着羧酸化反应时间的延长羧基含量和阻垢率快速增加,反应3 h时,羧酸化程度和阻垢率均达到最大;反应时间继续延长,羧酸化程度和阻垢率均在减小。这是因为反应时间过长会发生交联、成环等副反应,导致产物分子量增大,羧基含量减少。而羧基具有强大的螯合Ca2+的作用,能够显著增强阻垢效果,羧基含量大则阻垢效果好[20],羧基含量小则阻垢效果差。因此,最佳羧酸化反应时间为3 h。
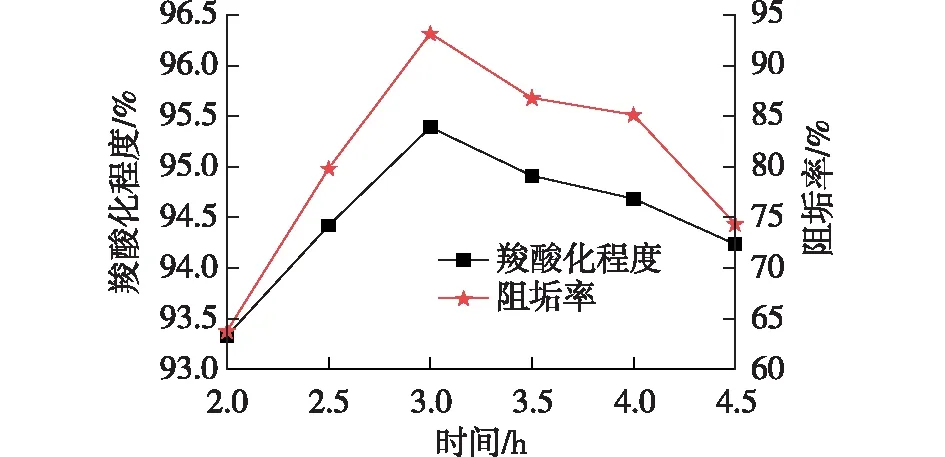
图6 羧酸化反应时间对羧酸化程度和阻垢率的影响Fig.6 Effects of carboxylation reaction time on carboxylation and scale inhibition rate
2.2 HBPE—OH与HBPE—COONa的FT-IR表征


图7 HBPE—OH和HBPE—COONa的FT-IR图谱Fig.7 FT-IR spectra of HBPE—OH and HBPE—COONa
2.3 NMR表征分析
2.3.1 核磁氢谱(1H NMR)表征分析
图8为HBPE—OH和HBPE—COONa的1H NMR图谱,由图8(a)所示,HBPE—OH图谱出现了4组峰,所表征的H位置分别是A为RCH2—OH、B为RC3—CH2—COOR、C为R3—CH2—OH、D为CH3—CR3,充分证明了HBPE—OH合成成功。对比图8(a)和8(b)发现:图8(b)中与酯基相连的亚甲基峰面积变大,说明酯基含量增多;与羟基相连的亚甲基峰面积变小,说明羟基含量变小;图8(b)中端基羧酸化后出现的一组大面积峰(A峰),此峰为酯基和羧基中间的亚甲基峰,说明有大量的羟基被成功修饰,催化剂DMAP性能优良。
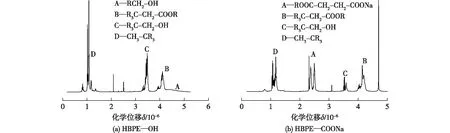
图8 HBPE—OH和HBPE—COONa的1H NMR图谱Fig.8 1H NMR spectra of HBPE—OH and HBPE—COONa
2.3.2 核磁碳谱(13C NMR)表征分析和支化度的确定
超支化结构由3种结构单元组成,分别为端基结构单元、线性结构单元和树形结构单元,如图9(a)所示,可通过核磁碳谱表征出来[21]。支化度是表征超支化聚酯的一个非常重要的参数,它能反映出超支化聚酯的结构特征,支化度DB计算公式见式(3)。
DB=(D+T)/(D+T+L)
(3)
式中:D、T和L分别为树形结构单元、端基结构单元和线性结构单元的峰面积。由图9(b)可以得到,端基结构单元、线性结构单元和树形结构单元的峰面积分别为1.00、1.72和0.25,根据式(3)计算得HBPE—OH的支化度为0.42,支化度较好。通过对比图9(b)和9(c)可以发现,端基羧酸化后端基结构单元消失,线性结构单元减少,而树形结构单元增多,支化度变为0.69,这是因为端基结构单元中的双羟基和线性结构单元中的单羟基被羧酸化而成为树形结构单元。仍然存在的少量羟基处于线性结构单元中,它被包裹在分子内部[22]。

图9 超支化聚酯结构单元和HBPE—OH及HBPE—COONa核磁碳谱Fig.9 Hyperbranched polyester structure unit and 13C NMR spectra of HBPE—OH and HBPE—COONa
2.4 CaSO4垢样表征分析
在不同质量浓度梯度阻垢剂下制得CaSO4垢样,通过扫描电子显微镜(SEM)表征结果如图10所示。由图10可见,不加阻垢剂形成的CaSO4晶体呈立方体棒状结构,棱角分明,表面光滑无缺陷,而加入阻垢剂后晶体尺寸明显变大,且表面不再完美,出现缺陷;随着阻垢剂质量浓度的增加,这一趋势更加严重,当阻垢剂质量浓度为7.5 mg/L时,CaSO4垢样表面犹如被刀割似的充满沟壑。由此可以明显看出,阻垢剂使CaSO4晶型发生了较大的改变,这是因为HBPE—COONa具有强大的吸附能力[23],阻垢剂分子会吸附在CaSO4的晶格阵列中占据本属于CaSO4晶胞的位置,使其不能按原来的堆砌方式规则地生长[24-26],导致其晶型改变,这就是阻垢剂的晶格畸变作用。

图10 不同质量浓度阻垢剂下CaSO4晶体SEM照片Fig.10 SEM images of CaSO4 crystals with different mass concentrations of scale inhibitors
3 结论
1)通过一锅熔融缩聚法合成了HBPE—OH,并进行端基羧酸化改性,得到了不含N、P元素的绿色HBPE—COONa阻垢剂。用单因素实验进行合成条件的优化,最终得到最佳缩聚时间4 h、缩聚温度140 ℃、缩聚催化剂质量分数为0.8%、羧酸化时间3 h、羧酸化温度70 ℃,且HBPE—COONa羧酸化改性程度较高,高达95.4%。
2)阻垢剂HBPE—COONa阻垢效果优于对比文献,当投加量质量浓度为3 mg/L时,阻垢率达到94%左右。
3)HBPE—OH和HBPE—COONa结构完整,支化度较好,充分证明了HBPE—OH合成成功,HBPE—COONa羧酸化程度高,说明DMAP对反应是一种高效的催化剂。
4)HBPE—COONa阻垢剂干扰了CaSO4成晶过程,使其由立方体棒状结构变得充满缺陷,证明了HBPE—COONa具有使CaSO4晶格畸变的作用。
猜你喜欢
分子催化(2022年1期)2022-11-02
建材发展导向(2021年16期)2021-10-12
建材发展导向(2021年7期)2021-07-16
科学与财富(2021年33期)2021-05-10
建材发展导向(2021年24期)2021-02-12
建材发展导向(2019年5期)2019-09-09
汽车零部件(2018年5期)2018-06-13
智富时代(2018年3期)2018-06-11
智富时代(2018年3期)2018-06-11
分析化学(2017年12期)2017-12-25
