
趋化因子和小胶质细胞在阿尔茨海默病神经炎症中的作用研究进展
2021-07-15 05:44:40佳张丽隗和儒翟悦怡刘树锋张连峰
中国比较医学杂志 2021年6期
王 佳张 丽隗和儒翟悦怡刘树锋*张连峰*
(1.河北省实验动物重点实验室河北医科大学,石家庄 050017;2.中国医学科学院医学实验动物研究所,北京协和医学院比较医学中心,卫计委人类疾病比较医学重点实验室,北京 100021)
阿尔茨海默病(Alzheimer disease,AD)是一种常见的慢性进行性神经退行性疾病,主要临床表现为进行性认知功能障碍、精神和行为异常,逐渐发展为无法进行日常生活的严重痴呆。 AD 主要病理学改变有大脑皮质区和海马区的细胞外β-淀粉样蛋白(amyloid β-protein,Aβ)沉积形成的老年斑(senile plaques,SP)、神经细胞内过度磷酸化的Tau蛋白错误折叠后聚集形成的神经原纤维缠结(neurofibrillary tangles,NFTs)、神经炎症和神经元丢失[1]。 目前,AD 的发病原因与发病机制尚不明确,经典的病因假说有Aβ 级联假说和Tau 蛋白假说等。 越来越多证据显示神经炎症在AD 发病中具重要作用,神经炎症是中枢神经系统(central nervous system,CNS)针对各种有害刺激(如损伤或感染)发生的免疫反应,由CNS 的神经胶质细胞、内皮细胞和外周的免疫细胞介导并产生细胞因子、趋化因子、活性氧等各种炎性介质共同引起[2]。 AD 病理研究发现SP 附近存在反应性小胶质细胞,同时检测到AD 病人脑实质中炎性细胞因子和趋化因子(chemokine,CK)的水平升高,揭示AD 中存在神经炎症,并参与AD 的发病机制[1,3]。 AD 中的神经炎症主要以Aβ 沉积和错误折叠的Tau 蛋白对小胶质细胞的持续激活,导致细胞因子和趋化因子等炎性介质的不断释放产生的慢性炎症反应,而释放的趋化因子诱导小胶质细胞向神经炎症区域迁移,发挥促炎或抗炎的作用,进而影响AD 的发展[4]。 因此,本文将重点论述小胶质细胞激活和趋化因子释放在AD 神经炎症中发挥的作用。
1 小胶质细胞的激活
小胶质细胞是神经胶质细胞的一种,大约占大脑中神经胶质细胞的10%左右。 研究表明小胶质细胞起源于卵黄囊的原始巨噬细胞,相当于脑和脊髓中的巨噬细胞[5]。 静息的小胶质细胞以休眠模式监视着周围组织的免疫状态,当出现炎症刺激时,小胶质细胞迅速被激活,通过改变形态迁移至病变部位,清除坏死物质,支持和保护神经系统。激活后的小胶质细胞可以极化为促炎或抗炎表型,分别称为经典激活小胶质细胞(M1 型)和替代激活小胶质细胞(M2 型)[6]。 M1 型为促炎状态,释放大量促炎因子(如 IL-1β、TNF-α、IFN-γ、CCL2)以及一氧化氮合酶iNOS,活性氧ROS 等炎性成分,不断加剧炎症反应,引起神经元变性及脑组织损伤。 M2型为抗炎状态,能够释放抗炎因子(如IL-4、IL-10、IL-13、YM-1)以及神经营养因子促进炎症消退,吞噬细胞碎片,促进组织修复并重建体内稳态[6-8]。
AD 产生的Aβ 沉积、错误折叠的Tau 蛋白及损伤的神经元会吸引小胶质细胞的聚集并引起其激活,同时引起细胞因子和趋化因子的释放,这些因素与Aβ 持续相互作用形成AD 中的神经炎症[9-10]。AD 病理研究中发现Aβ 可以诱导小胶质细胞的聚集并浸润在淀粉样斑块周围,Aβ 可以作为危险相关分子模式激活小胶质细胞表面模式识别受体如Toll 样受体(toll-like receptors,TLR),髓样细胞触发受体2(triggering receptor expressed on myeloid cells,TREM2),清道夫受体(scavenger receptor,SR-AI/II),补体受体,受体晚期糖基化终产物(receptor advanced glycosylation end product,RAGE)等,引起小胶质细胞的活化、分泌、吞噬等作用[11-12]。 Aβ 还可以直接以浓度依赖的方式与淀粉样前体蛋白(amyloid precursor protein,APP)相互作用,共同诱导小胶质细胞的激活并分泌炎性因子TNF-α[13]。Nussbaum 等[14]发现 Aβ 还能诱导 Tau 蛋白异常聚集,并在细胞内形成NFTs 而引发慢性神经炎症。相关研究报道在AD 患者海马体的NFTs 和带有缠结的神经元附近,以及Tau 蛋白转基因动物模型(TAUSHR72 转基因大鼠和 TauR406W 转基因小鼠)中,常出现小胶质细胞的激活,可能的原因是当Tau 蛋白发生错误折叠后,不断聚集并在神经元中形成NFTs,破坏神经元的功能并导致细胞最后死亡,引发的炎症激活小胶质细胞。 这些均表明炎症反应与NFTs 之间存在密切关系[10,15-16]。 同时激活的小胶质细胞释放的TNF-α 会在体外诱导Tau 蛋白的聚集[17]。 因此,Aβ 沉积和错误折叠的 Tau 蛋白可以通过多种途径激活小胶质细胞炎症反应路径,并进一步影响AD 的发生、发展。
静息的小胶质细胞在AD 产生的病理中可以诱导激活为M1 型和M2 型。 目前的研究认为在AD的早期,Aβ 沉积将静止的小胶质细胞激活为M2型,M2 型极化的小胶质细胞表现出神经保护和抗炎作用,分泌抗炎因子,吞噬、降解、去除Aβ 和Tau,抑制炎症反应。 随着 AD 病理发展,Aβ 和 Aβ 诱导的促炎因子持续相互作用使小胶质细胞过度活化转变为M1 型,M1 型的小胶质细胞表现出神经毒性和促炎作用,释放大量促炎因子,运动能力下降,吞噬、降解能力减弱,加剧炎症反应[18-19]。 图1 为小胶质细胞在AD 中的激活。 从图中发现,AD 中激活的M1 型小胶质细胞可以引起促炎性趋化因子(CCL2、CCL3、CCL4、CCL5、CXCL1、CXCL8、CXCL9、CXCL10)的分泌,M2 型小胶质细胞引起抗炎性趋化因子(CCL22、CXCL8、CXCL12、CX3CL1) 的分泌[20-23]。 由此可见,小胶质细胞的极化过程中会引起大量趋化因子的分泌,这些趋化因子与小胶质细胞相互作用,共同影响AD 的进程。 因此,下文将对趋化因子分类总结并分别展开论述。

图1 小胶质细胞在AD 中的激活过程Note. Deposition of Aβ, Tau protein, pathogens, apoptotic cell debris, etc. can all activate microglia in AD disease. After activation, microglia can be polarized into M1 and M2 types. M1 type microglia release CCL2, CCL3, CCL4, CCL5,CXCL1, CXCL8, CXCL9, CXCL10, IL-1α, IL-1β, IL-6, TNF-α, iNOS, and other pro-inflammatory factors; M2 type microglia release CCL22, CXCL8, CXCL12, CX3CL1, IL-4, IL-10, IL-13, TGF-β, FIZZ-1, YM-1, and other antiinflammatory factors. M1 type microglia recruit immune cells, activate the immune response, and further release inflammatory factors, leading to an inflammatory cascade, accelerating cell death and tissue damage. M2 type microglia phagocytize Aβ, clear cell debris, enhance neuroprotective effects, promote tissue repair, and maintain homeostasis.Figure 1 Activation process of microglia in AD
2 趋化因子的分泌
趋化因子是一类促使细胞分化、迁移和运输功能的多肽,能够激活趋化因子受体,在炎症过程中诱导趋化、组织外渗以及调节白细胞的功能[24]。 趋化因子及其受体在大脑中以低水平表达,受到炎症刺激才会发生调节作用,其表达主要来源于小胶质细胞及其他神经细胞[25]。 趋化因子根据分子中N-末端半胱氨酸的不同位置分为四个亚家族,包括CXC、CC、C 和 CX3C。 其中 CXC 趋化因子亚族 17个成员,C 趋化因子亚族2 个成员,CX3C 趋化因子亚族1 个成员。 而CC 趋化因子则是趋化因子家族中最大的亚类,包括 28 个成员,分别为 CCL1 ~CCL28。 CXC 趋化因子家族成员中 CXCL1、CXCL9、CXCL10 在 AD 中上调,参与促炎反应[22-23]。 CX3C家族的唯一成员CX3CL1 发现具有抑制Tau 蛋白病理改变,增加神经信号传导和神经保护作用[26]。 更多的研究发现大量的CC 趋化因子在AD 中上调,少量因子存在下调或者不变[27],且其受体 CCR3,CCR5 阳性反应的小胶质细胞与 Aβ 沉积密切相关[28]。 提示了CC 趋化因子在AD 中可能的重要作用和未来研究方向。 因此,本文重点论述CC 趋化因子在AD 神经炎症和小胶质细胞的激活中的重要作用。 28 个CC 趋化因子的受体、功能和在AD 中的表达变化总结见表1。 根据28 个CC 趋化因子目前已发现的炎症调节作用将其分为三类:促炎性、抗炎性和双重功能的趋化因子。 分述见表1。
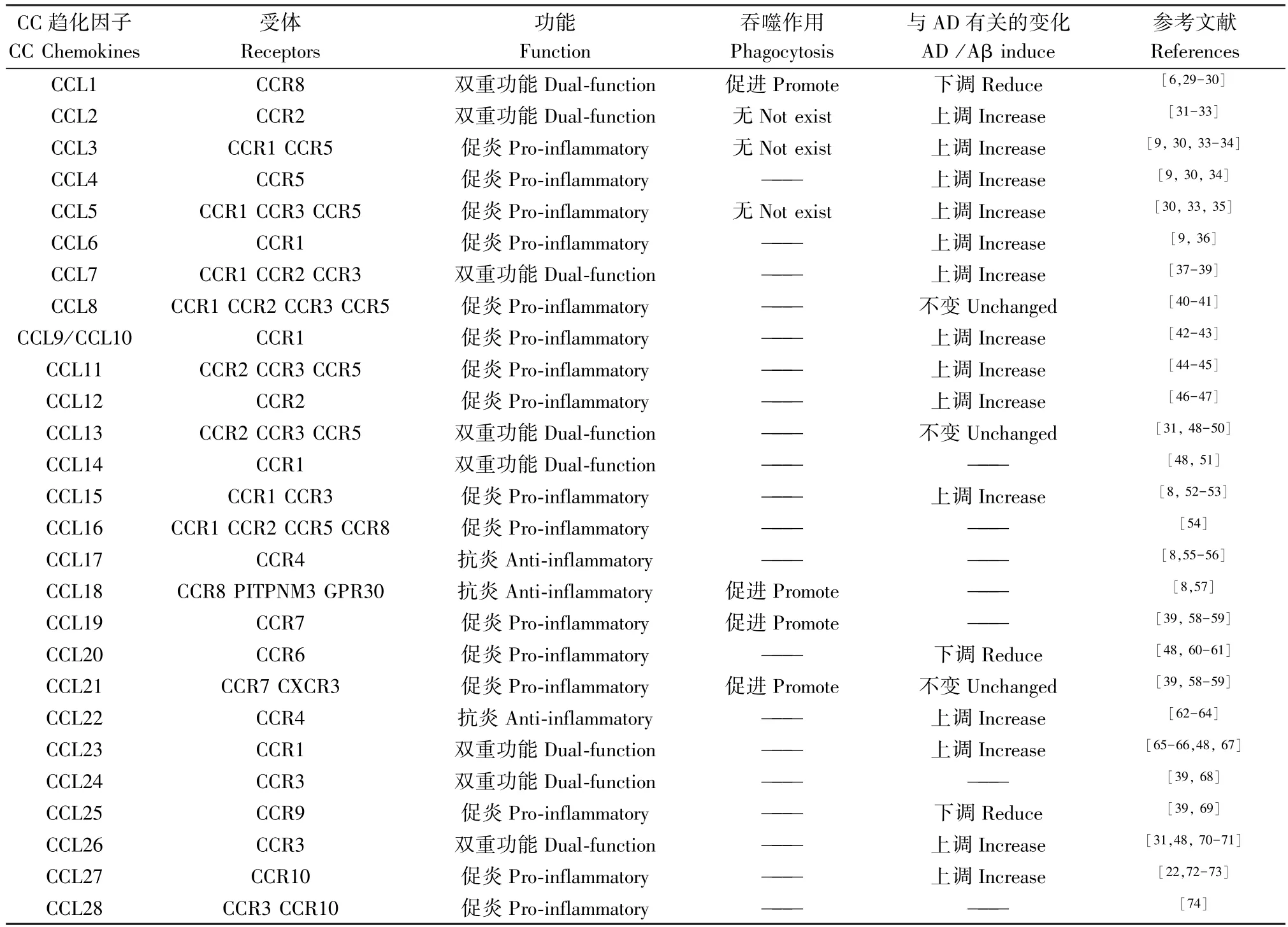
表1 CC 趋化因子的功能及其受体Table1 CC Chemokines function and its receptors
2.1 促炎性趋化因子
促炎因子对炎症的发展有促进作用。 大多数表现出促炎作用的趋化因子在AD 及其产生的神经炎症中是上调的,如表 1 中 CCL2、CCL3、CCL4、CCL5、CCL6、CCL9、CCL11、CCL12、CCL15,这些趋化因子不单表现参与炎症反应,也在AD 的病理机制中发挥特有的作用,并且和其本身的氨基酸序列同源性密切相关。
CCL2,又称为单核细胞趋化蛋白(monocyte chemoattractant protein,MCP-1),由淀粉样斑块相关的小胶质细胞产生,Kiyota 等[75]的研究发现CCL2过表达的Tg2576(APPswe)/CCL2 转基因小鼠表现出小胶质细胞的聚集和促进Aβ 沉积和淀粉样斑块形成,并加速了认知障碍。 CCL2 过表达使rTg4510(tauP301L)转基因小鼠模型的Tau 蛋白病理恶化,表现以NFTs 和磷酸化Tau 阳性包涵体的大量增加,并伴有胶质细胞增生和明显的炎症反应[76]。 另一研究表明CCL2 在遗忘性轻度认知障碍(amnestic mild cognitive impairment,aMCI)、AD 及同样具有痴呆、脑萎缩特征的额颞叶型失智症(frontotemporal dementia,FTLD)患者的脑脊液(cerebrospinal fluid,CSF)中均明显升高[77-78]。 CCL12(MCP-5) 是与CCL2(MCP-1)同源的单核细胞趋化因子,具有66%的氨基酸同一性。 CCL2 与CCL12 都是Tau 病理相关神经炎症的压力应激反应基因[46]。 CCL2 和CCL12 还可以同时与CCR2 受体结合,CCR2 在小胶质细胞上具有迅速促进嘌呤能受体(purinergic receptor,P2RX4)转运到细胞表面的能力,进而促进小胶质细胞的胞吐作用[79]。 脊髓中星形胶质细胞表达的CCL7(MCP-3)和CCL2(MCP-1)具有大于60%的氨基酸同一性,也可以通过CCR2 激活小胶质细胞,产生更多炎性介质引起神经性疼痛[37,80]。根据以上证据说明具有同源性基因的趋化因子可以与同一种受体结合,对炎症刺激发挥同样的促炎功能。
然而这一现象并不是完全一致的,有研究报道在212 名FTLD 患者与203 名年龄匹配的对照人群观察 CCL2(MCP-1)A-2518G 的单核苷酸多态性(single nucleotide polymorphism,SNP),FTLD 患者脑脊液中 MCP-1 水平显著高于对照组,MCP-1 A-2518G SNP 可能通过影响 MCP-1 的产生而成为FTLD 的保护因子[81]。 CCL2 和 CCL8(MCP-2)同样具有相似序列的趋化因子,其氨基酸序列同源性为62%,均会在神经退行性疾病中升高[80]。 在CCL8中发现 SNP 与 CCL2 都位于同一连锁区,其中rs1163763 会导致氨基酸的取代,对蛋白质功能产生潜在的影响,对219 名AD 患者和209 名FTLD 患者进行了rs1133763 关联测试,并与231 名年龄相匹配的对照组进行比较,发现CCL8 rs1133763 的分布在患者和对照组之间没有显着差异。 这种SNP 相关的连锁不平衡基因变异对神经退行性疾病并无明显作用[40]。 因此,即使是具有同源性基因的趋化因子在神经疾病中也会发挥各自不同的功能。
CCL3/巨 噬 细 胞 炎 性 蛋 白-1α (Macrophage inflammatory protein,MIP-1α)和 CCL4/巨噬细胞炎性蛋白-1β(Macrophage inflammatory protein,MIP-1β)是巨噬细胞炎性蛋白(MIP-1)的两种形式,二者相互作用,共用同一受体CCR5,其功能与炎症反应有关[82]。 5XFAD 转基因小鼠中淀粉样斑块相关的小胶质细胞表现出免疫反应过度和炎症的高反应性,同时发现促炎因子 CCL3、CCL4、CCL6 的表达[9]。 Passos 等[83]的研究也发现在小鼠侧脑室注射Aβ1-40 后,CCL3 及其受体CCR5 的表达水平升高,小胶质细胞的数量也显著增加。 遗传方面CCL3/MIP-1α 的基因多态性影响中国人对AD 的敏感性,其中MIP-1α-906(TA)6/(TA)6 基因可能是AD 的遗传危险因素[84]。 研究发现AD 患者的外周血单核细胞控制着 CCL4 的产生[85], AD 的APPswe/PS1dE9 转基因小鼠大脑的CCL4 水平升高与大脑Aβ 沉积呈年龄依赖性相关,并增加淀粉样斑块周围星形胶质细胞的活化,放大了炎症反应[86]。
2.2 抗炎性趋化因子
抗炎因子被认为可以减轻炎症反应。 目前明确具有抗炎作用的趋化因子CCL17、CCL18、CCL22在有关AD 疾病中的研究较少,但在影响大脑认知、神经炎症、小胶质细胞的激活等神经系统相关研究中发现了很多可能会影响AD 疾病发展的抗炎作用。
CCL17,即胸腺和激活调节趋化因子(thymus and activation-regulated chemokine,TARC),是 M2 型巨噬细胞的标志物,IL-4 可以诱导其在巨噬细胞中形成和上调,激活的M2 型巨噬细胞参与吞噬细胞碎片以及抑制炎症反应[55]。 维生素D3 可选择性地增强小胶质细胞HMO6 中细胞因子IL-10 和CCL17的表达,使小胶质细胞具有抗炎活性,保护神经进行免疫修复[56]。 在应对LPS 诱导的急性炎症刺激时,与记忆认知有关的海马CA1 区小胶质细胞的CCL17 上调,CCL17 可以维持小胶质细胞的静息状态,CCL17 基因敲除小鼠(CCL17-/-)的小胶质细胞体积减少,并呈现出分枝减少、极性增强的反应形态[87]。 一项针对高罹患AD 疾病风险的墨西哥裔美国人关于遗忘性轻度认知障碍的生物标记物检测发现在aMCI 病例中血液生物标记物以炎性因子为主,排列前三的标记物分别为TNFα,IL-10 和TARC,这些发现提示了炎性因子和aMCI 发展至AD 的代谢过程可能存在相互作用,还需要对上述炎性因子进一步研究[88]。
CCR4 是 CCL17 和 CCL22/巨噬细胞来源的趋化因子(macrophage-derived chemokine,MDC)的共同受体,CCR4 的基因敲除小鼠(CCR4-/-)表现出运动和探索行为受损,此时的 CCR4 与其配体CCL22 可能参与了神经元和神经胶质细胞的功能调节[89]。 CCL22 在实验性自身免疫性脑脊髓炎(experimental autoimmune encephalomyelitis,EAE)小鼠的大脑中由小胶质细胞产生,通过诱导TH2 细胞的归巢来调节 Th1 细胞介导的神经炎症[90]。CCL22 同样具有抗炎活性,在神经系统疾病的脱髓鞘、神经元损伤中,都检测到了M2 型小胶质细胞标志物 IL-10、CCL18、CCL22,激活的 M2 型小胶质细胞有助于免疫抑制,促进神经修复和髓鞘的再生[91]。 Movsesyan 等 的 研 究 团 队[92-93]设 计 以CCL22 作为分子佐剂的 AD 疫苗 PMDC-3Aβ1-11-PADRE,通过诱发细胞免疫及体液免疫,产生抗炎作用,减轻炎症反应,协同疫苗产生的抗Aβ 抗体共同促进APPSwe/PS1M146V/tauP301L 转基因小鼠大脑中Aβ 沉积物的清除,抑制Aβ 病理学的积累。
CCL18 也称为替代巨噬细胞活化相关趋化因子- 1 (alternative macrophage activation associated chemokine,AMAC-1)和巨噬细胞炎性蛋白-4(MIP-4),它与CCL3 关系最密切,共享64%的序列同一性,却没有激活与CCL3 相同的受体,因为CCL18 具有独特的四级结构,可以和CCR8、PITPNM3、GPR30三种受体结合,表现出抗炎性趋化因子的作用。CCL18 作为人和灵长类动物独有的趋化因子,从死亡后人脑组织分离出来的小胶质细胞在IL-4 刺激下培养,发现了 CCL18 上调[94]。 在没有 IL-4 的刺激,CCL18 也可以诱导单核细胞成为M2 型巨噬细胞,上调抗炎因子IL-10,并增强巨噬细胞的吞噬能力,清除细胞碎片[57]。
2.3 双重功能趋化因子
有学者在炎症相关研究中发现个别趋化因子具有促炎和抗炎的双重作用,即在不同的炎症环境中可以表现出促炎状态也可以表现出抗炎状态,如CCL1、CCL2、CCL7、CCL13、CCL14、CCL23、CCL24、CCL26。 CCL1、CCL2、CCL7 在神经炎症中表现出以促炎作用为主,而另外一些具有双重功能的趋化因子在AD 和神经炎症中的作用并不十分明确。
CCL23 是具有促炎和抗炎双重功能的趋化因子,在单核细胞中既能被IL-1β 和IFN-γ 诱导表达,也能由IL-4 和IL-13 诱导表达,在树突状细胞由IL-10 诱导使其表达[65]。 临床研究表明AD 患者血液中的 CCL23 高于健康对照者,从轻度认知障碍(mild cognitive impaired,MCI)发展到AD 患者的血液及脑脊液检测数据中发现CCL23 呈高进展性,并且在AD 遗传易感因素ApoE ε4 等位基因携带者血液中检测到高水平的CCL23,可能与血浆中的炎症反应有关,预测CCL23 可能是轻度认知障碍发展到AD 的血液生物炎性标志物[66]。 CCL23 的受体CCR1,只在与Aβ42 阳性的神经炎斑和营养不良性神经元中表达,并且随临床疾病的严重程度而增加,因此成为AD 特有的神经炎性标志物[95]。
CCL26 又称嗜酸性粒细胞趋化因子(eotaxin-3),已证明 IL-4 和 IL-13 可通过 JAK1-STAT6 途径上调CCL26 的表达,表现出抗炎作用,但同时TNF-α 对 IL-4 增强的 CCL26 产生协同作用[96]。 在 EAE大鼠的神经炎症反应中,CCL26 结合CCR3 发挥促炎作用,加重脑组织损伤[70]。 在轻度认知障碍发展至AD 的临床随访研究中发现前驱性AD 患者脑脊液中的CCL26 显著高于健康对照者[31]。
3 小结与展望
无论是衰老、遗传或者环境因素造成的痴呆,都会在AD 病理形成的过程中产生神经炎症。 小胶质细胞是大脑中免疫监视器,也是神经炎症反应的核心。 小胶质细胞作为AD 清除Aβ 的主要途径,可以抑制淀粉样蛋白的沉积延缓AD 的发展;但炎性持续激活的小胶质细胞会分泌更多的促炎因子,引起神经元的损伤,加速AD 的进展。 小胶质细胞是AD 疾病发展的一把双刃剑,如何抑制M1 型小胶质细胞的炎性激活,减轻炎症反应,增加M2 型小胶质细胞的神经保护作用,维持稳态的正性平衡成为治疗AD 疾病的方向。
趋化因子在AD 的神经炎症反应中具有双向调节的作用,一方面具有促炎作用的趋化因子可以持续激活M1 型小胶质细胞分泌更多炎性因子及毒性物质,使小胶质细胞失控,加重炎症反应,形成恶性循环;另一方具有抗炎作用的趋化因子可以维持M2型小胶质细胞的稳态,增加Aβ 内化及降解,减轻炎症的活跃程度。 这是一个高度动态的过程,神经炎症中的促炎和抗炎因子对于正常细胞组织代谢的动态平衡至关重要,如何维持稳态平衡决定了炎症反应的发展。 AD 疾病中的神经炎症活跃程度由趋化因子、细胞因子等炎性介质所反应,抗炎因子和促炎因子的平衡影响着AD 的预后。 很多研究将促炎性趋化因子CCL2、CCL3 作为MCI 发展到 AD 早期的炎性因子标志物,也有将具有抗炎作用的CCL22 作为分子佐剂的AD 疫苗,但更多关于趋化因子的研究只是检测其在AD 中的水平变化,并没有深入探索这些趋化因子对于小胶质细胞或者Aβ诱导神经炎症的作用机制研究。 因此需要明确抗炎性趋化因子控制小胶质细胞表型转换的机制,进而可以通过增加抗炎性趋化因子的正向作用和减少促炎性趋化因子的负向作用来改善AD 的神经炎症程度,为AD 的治疗提供新的思路。
猜你喜欢
神经损伤与功能重建(2020年11期)2020-12-01 05:01:54
中华养生保健(2020年7期)2020-11-16 01:13:32
中成药(2017年9期)2017-12-19 13:34:20
中成药(2017年10期)2017-11-16 00:50:09
湖南中医药大学学报(2016年1期)2016-12-01 04:08:21
中国医药生物技术(2015年4期)2015-12-26 08:26:34
磁共振成像(2015年1期)2015-12-23 08:52:21
中药与临床(2015年5期)2015-12-17 02:39:30
现代检验医学杂志(2015年6期)2015-02-06 01:44:04
河南医学研究(2014年4期)2014-02-27 14:52:17
