
以橙皮苷为对照品的内加入法测定甘草中甘草苷和甘草酸含量
2021-07-15 01:27余敏灵
中国药业 2021年13期
田 涛,余敏灵
(1. 成都储供基地成都药材供应站,四川 成都 610017; 2. 四川省乐山市食品药品检验检测中心,四川 乐山 614000)
甘草为豆科植物甘草 Glycyrrhiza uralensis Fisch.、胀果甘草 Glycyrrhiza inflata Bat. 或光果甘草 Glycyrrhizaglabra L. 的干燥根和根茎,收载于 2015 年版《中国药典(一部)》,具有补脾益气、清热解毒、祛痰止咳等功效[1-3]。采用以甘草酸铵、甘草苷为对照品的外标法测定甘草酸和甘草苷含量[4],存在对照品价格昂贵、易吸潮等缺陷[5]。替代对照品法是解决该问题的主要方法[6-13]。本研究中采用替代对照品内加入法,以橙皮苷、甘草苷、甘草酸铵色谱峰保留时间和紫外光谱特征相结合的定性方法,将对照品色谱峰提取的紫外光谱图所建立的纯度检查图库应用于色谱峰的定性中,通过甘草苷和甘草酸铵相对橙皮苷的相对校正因子( f)计算甘草中甘草苷和甘草酸的含量,并对3 批样品的测定结果与法定标准测定数据进行比较。现报道如下。
1 仪器与试药
1.1 仪器
Agilent 1260 型高效液相色谱仪(美国安捷伦公司),配有二极管阵列检测器和OpenLAB CDS 工作站;PE-A10 型高效液相色谱仪(珀金埃尔默公司),配有二极管阵列检测器;Ultimate 3000 型高效液相色谱仪(赛默飞世尔公司),配有二极管阵列检测器;XSE205 Dual-Range 型电子天平(梅特勒-托利多公司,精度为0.01mg);ULUP-Ⅱ-10T 型优普系列超纯水器(成都超纯科技有限公司);CQ-100B 型超声仪(上海跃进医用光学器械厂,功率为200 W,频率为40 kHz)。
1.2 试药
甘草苷对照品(批号为111610-201607,含量为93.1%),甘草酸铵对照品(批号为 110731 -201720,含量为97.7%),橙皮苷对照品(批号为110721-201818,含量为96.2%),均购于中国食品药品检定研究院;甘草酸单铵盐A 原料药(北京凯因科技股份有限公司,批号为1707003,含量为67.2%);甘草(乐山市康发药业有限公司,批号分别为 180913311,190613311,180721311);乙腈为色谱纯,磷酸、甲醇、三乙胺等均为分析纯。
2 方法与结果
2.1 色谱条件
色谱柱:Welch Ultimate LP-C18柱(250 mm×4.6 mm,5 μm);流动相:乙腈(A)- 0.1% 三乙胺溶液(用磷酸调节 pH 至 2.5,B),梯度洗脱 (程序见表 1);流速:1.0 mL /min;检测波长:276 nm(甘草苷),284 nm(橙皮苷),252 nm(甘草酸铵);进样量:10 μL;柱温:30 ℃。
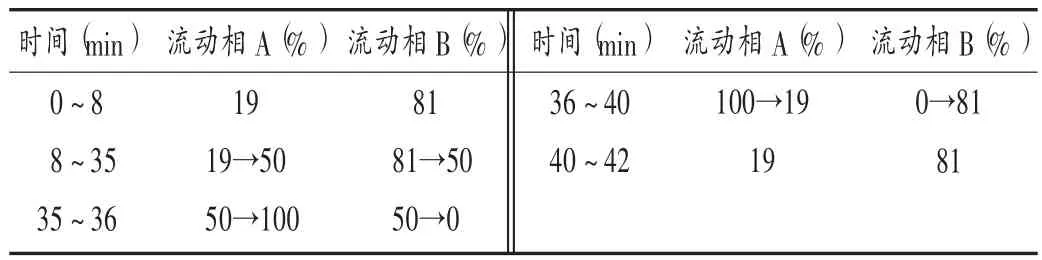
表1 流动相梯度洗脱程序Tab.1 Gradient elution procedure of mobile phase
2.2 溶液制备
混合对照品溶液:取甘草苷对照品17.84 mg,精密称定,置100 mL 容量瓶中,用80%甲醇溶液溶解并稀释至刻度,摇匀,作为甘草苷对照品贮备液;取橙皮苷对照品 64.12 mg,精密称定,置250 mL 容量瓶中,加甲醇适量溶解并稀释至刻度,摇匀,作为橙皮苷对照品贮备液;取甘草酸铵对照品11.08 mg,精密称定,置10 mL容量瓶中,用80%甲醇溶液溶解并稀释至刻度,摇匀,作为甘草酸铵对照品贮备液。分别精密量取甘草苷对照品贮备液2.5 mL,橙皮苷对照品贮备液2 mL,甘草酸铵对照品贮备液1 mL,置同一10 mL 容量瓶中,加80%甲醇溶液稀释至刻度,摇匀,即得。
供试品溶液:取甘草(批号为180913311)粉碎,过3号筛,取细粉 0.2 g,精密称定,置 50 mL 容量瓶中,加80%甲醇溶液适量,超声处理(功率为250 W,频率为40 kHz)30 min,放冷,加80%甲醇溶液稀释至刻度,摇匀,即得。
橙皮苷-样品混合液:另取上述细粉0.2 g,精密称定,置50 mL 容量瓶中,加80%甲醇溶液适量,超声处理(功率为250 W,频率为 40 kHz)30 min,放冷,精密加入橙皮苷对照品贮备液10 mL,用80%甲醇溶液稀释至刻度,摇匀,即得。
空白溶剂:80%甲醇溶液。
2.3 测定波长选择
在混合对照品溶液提取的甘草苷、橙皮苷、甘草酸铵紫外光谱图中,甘草苷、橙皮苷、甘草酸铵分别在276,284,252 nm 波长处有最大吸收,光谱图见图 1。故选择测定波长为276 nm(甘草苷)、284 nm(橙皮苷)、252 nm(甘草酸铵)。
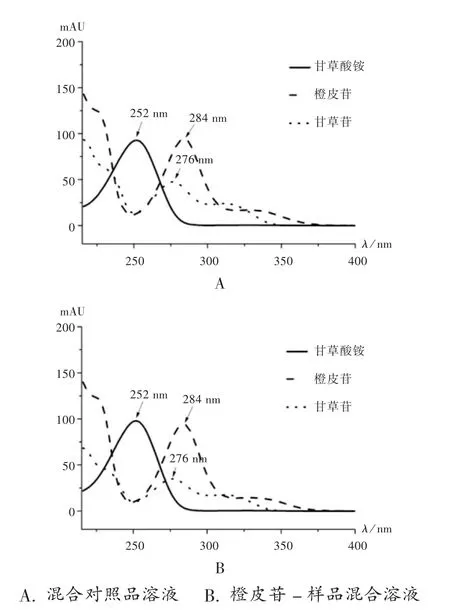
图1 紫外光谱图A.Mixed reference solution B.Hesperidin-sample mixed solutionFig.1 Ultraviolet spectrogram
2.4 f 耐受性考察
取2.2 项下混合对照品溶液适量,按2.1 项下色谱条件进样测定,考察耐受性[14-15]。分别在使用 3 台不同型号液相色谱仪(Aglient 1260 型、PE -A10 型、Ultimate 3000 型 ),测 定 波 长 (248 /272 /280,250 /274 /282,252 /276 /284,254 /278 /286,256 /280 /288 nm),柱温(28.0,29.0,30.0,31.0,32.0℃),流动相 pH 值(2.3,2.5,2.7,2.9),流速(0.900,0.950,1.000,1.050,1.100mL/min)条件下测定,计算 f。结果见表2。

表2 相对校正因子耐受性考察结果Tab.2 Results of tolerance test of relative correction factors
2.5 f 测定
分别取 2.2 项下混合对照品溶液 2,5,10,15,20,25μL,按2.1 项下色谱条件测定,记录色谱图。以峰面积(Y)为纵坐标,以进样量(X,μg)为横坐标进行线性回归(校正截距为 0)。回归方程分别为 Y甘草苷=1 929.190 8 X甘草苷(r =0.999 5)、Y橙皮苷=1 783.181 4 X橙皮苷(r =0.999 4)、Y甘草酸铵=802.771 3 X甘草酸铵(r =0.999 7)。结果表明,甘草苷、橙皮苷和甘草酸铵进样量分别在0.083 0 ~1.038 1 μg、0.098 7 ~ 1.233 7 μg、0.216 5 ~ 2.706 3 μg范围内与峰面积线性关系良好。以甘草苷和甘草酸铵回归曲线斜率与橙皮苷回归曲线斜率的比值分别计算 f,以2.4 项下3 台液相色谱仪所测平均值为最终 f,结果甘草苷、甘草酸铵与橙皮苷在不同考察条件下测得的 f的平均值分别为 1.081 3 和 0.449 5,RSD 分别为0.27%和 0.23%(n =3)。
2.6 紫外光谱稳定性和相对保留时间考察
取2.2 项下混合对照品溶液和橙皮苷-样品混合溶液,按2.1 项下色谱条件进样测定,除测定波长外,按2.4项下条件测定,记录色谱图。比较混合对照品溶液和橙皮苷-样品混合溶液各组分在不同条件下色谱峰相对保留时间的平均值和相对标准偏差、提取紫外光谱图中吸收曲线的最大吸收波长及各组分对照品建立的紫外图库比较的匹配因子平均值,结果见表3。可见,不同测定条件下相对保留时间变化的 RSD 均未超过规定值2.0%,在各测定条件下,甘草苷(276 nm)/橙皮苷(284 nm)/甘草酸铵(252 nm)最大吸收波长均无变化(≤±2 nm),匹配因子均大于 999.96 /1 000。

表3 紫外光谱稳定性和相对保留时间耐受性考察结果Tab.3 Results of UV spectrum stability test and relative retention time tolerance test
2.7 方法学考察
专属性试验:分别取2.2 项下混合对照品溶液、供试品溶液、橙皮苷-样品混合溶液、空白溶剂,按2.1 项下色谱条件测定,色谱图见图2。结果橙皮苷-样品混合溶液色谱图中,有与混合对照品溶液色谱图中甘草苷、橙皮苷和甘草酸铵相应的色谱峰,各组分色谱峰与其相邻的色谱峰分离度均大于1.5;供试品溶液色谱图中,有与混合对照品溶液色谱图中甘草苷和甘草酸铵相应的色谱峰,且样品溶液色谱图中,在与橙皮苷-样品混合溶液色谱图橙皮苷色谱峰相应位置上无色谱峰。以混合对照品溶液色谱图中橙皮苷、甘草苷、甘草酸铵的色谱峰提取紫外图谱建立其相应的纯度检查图库,并与橙皮苷-样品混合溶液色谱图中各成分色谱峰紫外图谱比较,橙皮苷色谱峰匹配因子为999.954/1 000,甘草苷色谱峰匹配因子为999.917/1 000,甘草酸铵匹配因子为999.936/1 000。橙皮苷色谱峰与甘草苷和甘草酸铵色谱峰保留时间的比值分别为1.335 0 和0.541 0。由图2 C 可知,混合对照品溶液色谱图中,甘草苷、橙皮苷、甘草酸铵出峰时间分别为13.213,17.639,32.602min;混合对照品溶液和橙皮苷-样品混合溶液色谱图中,橙皮苷、甘草苷、甘草酸铵色谱峰紫外光谱图分别于284,276,252 nm 波长处有最大吸收。
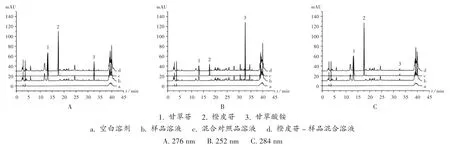
图2 高效液相色谱图1. liquirtin 2.hesperidin 3.ammonium glycyrrhizinatea.Blank solution b.Sample solution c.Mixed reference solution d. Hesperidin - sample mixed solutionA.276 nm B.252 nm C.284 nmFig.2 HPLC chromatograms
精密度试验:取2.2 项下混合对照品溶液,按2.1项下色谱条件重复进样6 次,记录色谱图。结果甘草苷、橙皮苷、甘草酸铵的 RSD 分别为 0.22% ,0.18% ,0.23%(n =6),表明仪器精密度良好。
重复性试验:取 2.2 项下甘草细粉 0.2 g,精密称定,共6 份,依法制备橙皮苷-样品混合溶液,按2.1 项下色谱条件进样测定,记录色谱图。结果橙皮苷平均峰面积为881.4,甘草苷和甘草酸铵的平均峰面积(以称样量为 0.2 g 计算)分别为 562.4 和 817.5,RSD 分别为 0.65%和 0.72%(n =6),表明方法重复性好。
稳定性试验:取2.2 项下橙皮苷-样品混合溶液,分别于室温放置 0,2,4,6,8,12,24 h 时,按 2.1 项下色谱条件进样测定。结果甘草苷、橙皮苷和甘草酸铵色谱峰峰面积变化不大,RSD 分别为 0.28% ,0.37% ,0.31%(n =7),表明供试品溶液在室温下放置24 h 内稳定。
检测限与定量限确定:取2.2 项下混合对照品溶液1 mL,置25 mL 容量瓶中,加80%甲醇溶液稀释至刻度,摇匀,作为供试品溶液,按2.1 项下色谱条件测定,记录色谱图。结果甘草苷(276 nm)、甘草酸铵(252 nm)的信噪比(S / N)分别为 30.4 和 77.6。分别以 S / N 的3 倍和10 倍计算检测限和定量限,结果甘草苷(276 nm)的检测限与定量限(以进样量计)分别为1.64 ng 和5.46 ng,甘草酸铵(252 nm)的检测限与定量限(以进样量计)分别为 1.67 ng 和 5.58 ng。
加样回收试验:取甘草酸单铵盐A 原料药334.82mg,精密称定,置100 mL 容量瓶中,用80%甲醇溶液溶解并稀释至刻度,摇匀,作为甘草酸铵原料药贮备液;取2.2 项下甘草细粉(每 1 g 样品中含甘草苷 7.296 1 mg,甘草酸 24.914 1 mg)0.1 g,精密称定,置 50 mL 容量瓶中,精密加入甘草苷对照品贮备液4 mL,甘草酸铵原料药贮备液1 mL,橙皮苷对照品贮备液10 mL,按2.2 项下方法制备橙皮苷-样品混合溶液,平行6 份,按2.1项下色谱条件进样测定,记录色谱图。按 Cm=(Am×Cs)/(f× AS)计算含量。式中,Cm为供试品溶液中甘草苷或甘草酸铵的进样量(μg),Am为供试品溶液中甘草苷或甘草酸铵的峰面积,Cs与 AS分别为供试品溶液中橙皮苷的进样量(μg)与峰面积。甘草酸含量=测得的甘草酸铵含量×0.979 7。结果见表4。
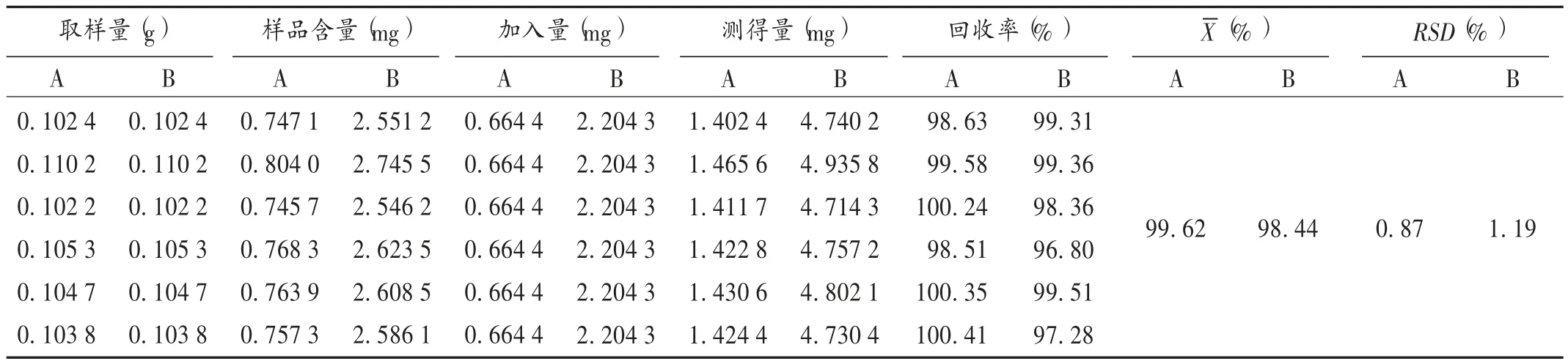
表4 替代对照品法加样回收试验结果(n =6)Tab.4 Results of the recovery test by the replacement method of chemical reference substance(n =6)
2.8 样品含量测定
取甘草,粉碎,过3 号筛,取细粉0.2 g,精密称定,置50 mL 容量瓶中,依法制备橙皮苷-样品混合溶液和供试品溶液,取橙皮苷-样品混合溶液,按2.1 项下色谱条件进样测定,记录色谱图。按加样回收试验项下方法计算被测定样品中甘草苷和甘草酸铵进样量,以进样量计算每1 g 样品中甘草苷和甘草酸含量,并将测定结果与法定检验方法测定结果比较。结果见表5。

表5 3 批样品含量测定结果Tab.5 Content determination of liquirtin and glycyrrhizic acid in three batches of samples
3 讨论
替代对照品内加入法是将替代对照品定量加入被测定溶液中作为供试品溶液,并按规定色谱条件进样分析,以色谱图中替代对照品峰面积和被测定成分峰面积,通过替代对照品与被测定成分的 f 计算被测定成分含量的方法。使用替代对照品内加入法测定样品,应对未加入替代对照品的样品溶液进行分析,在替代对照品所选定的检测波长的色谱图中,与替代对照品保留时间一致的位置上不应有其他干扰峰。
本试验中,色谱柱的改变对相对保留时间有较大影响,建议实验室采用Welch Ultimate LP-C18柱(250 mm×4.6 mm,5 μm);定性色谱峰时,参照国家药品监督管理局标准YBH06492018 有关物质项下相对保留时间确定方法,建议相对保留时间的数值保留一位有效数字,通过与对照品色谱峰所建立的紫外纯度检测图库比较,并按匹配因子和紫外特征吸收进行定性,由于在OpenLAB CDS 工作站建立的图库文件只能应用于其他Agilent 高效液相色谱仪,二极管阵列检测器和系列OpenLAB CDS 工作站(安捷伦公司),故采用该法作为色谱峰定性使用有较大局限性,在其他操作系统应用时还需建立其他操作系统的相关图库。
替代对照品内加入法中,不再单独制备和测定对照品溶液,简化了操作过程。该方法同时测定对照品和样品,与外标法相比,结果更准确,测定时间更短,相对保留时间更稳定。但由于替代对照品是加入供试品溶液,试验时要确认样品溶液色谱图在替代对照品色谱峰的位置是否有其他物质色谱峰的干扰,以排除干扰,可采用供试品溶液色谱图和样品加替代对照品溶液色谱图比较或色谱峰纯度检查。
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
中国应急管理科学(2021年4期)2021-04-13
中国合理用药探索(2021年2期)2021-01-03
中西医结合心血管病电子杂志(2020年23期)2020-09-26
临床医药文献杂志(电子版)(2020年43期)2020-07-30
中学生数理化·高一版(2020年9期)2020-01-02
中学化学(2019年4期)2019-08-06
考试周刊(2018年68期)2018-09-17
幸福·健康版(2018年3期)2018-03-23
食品与健康(2017年9期)2017-09-13
