
2D/2D Black Phosphorus/g-C3N4 S-Scheme Heterojunction Photocatalysts for CO2 Reduction Investigated using DFT Calculations
2021-07-13 09:53XingangFeiHaiyanTanBeiChengBichengZhuLiuyangZhang
物理化学学报 2021年6期
Xingang Fei , Haiyan Tan , Bei Cheng , Bicheng Zhu ,*, Liuyang Zhang ,*
1 State Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan 430070, China.
2 School of Chemistry and Environmental Engineering, Hubei University for Nationalities, Enshi 445000, Hubei Province, China.
Abstract: Photocatalytic reduction of CO2 to hydrocarbon compounds is a promising method for addressing energy shortages and environmental pollution.Considerable efforts have been devoted to exploring valid strategies to enhance photocatalytic efficiency. Among various modification methods, the hybridization of different photocatalysts is effective for addressing the shortcomings of a single photocatalyst and enhancing its CO2 reduction performance. In addition, metal-free materials such as g-C3N4 and black phosphorus (BP) are attractive because of their unique structures and electronic properties. Many experimental results have verified the superior photocatalytic activity of a BP/g-C3N4 composite. However, theoretical understanding of the intrinsic mechanism of the activity enhancement is still lacking. Herein, the geometric structures, optical absorption, electronic properties, and CO2 reduction reaction processes of 2D/2D BP/g-C3N4 composite models are investigated using density functional theory calculations. The composite model consists of a monolayer of BP and a tri-s-triazine-based monolayer of g-C3N4. Based on the calculated work function, it is inferred that electrons transfer from g-C3N4 to BP owing to the higher Fermi level of g-C3N4 compared with that of BP. Furthermore, the charge density difference suggests the formation of a built-in electric field at the interface, which is conducive to the separation of photogenerated electron-hole pairs. The optical absorption coefficient demonstrates that the light absorption of the composite is significantly higher than that of its singlecomponent counterpart. Integrated analysis of the band edge potential and interfacial electronic interaction indicates that the migration of photogenerated charge carriers in the BP/g-C3N4 hybrid follows the S-scheme photocatalytic mechanism.Under visible-light irradiation, the photogenerated electrons on BP recombine with the photogenerated holes on g-C3N4,leaving photogenerated electrons and holes in the conduction band of g-C3N4 and the valence band of BP, respectively.Compared with pristine g-C3N4, this S-scheme heterojunction allows efficient separation of photogenerated charge carriers while effectively preserving strong redox abilities. Additionally, the possible reaction path for CO2 reduction on g-C3N4 and BP/g-C3N4 is discussed by computing the free energy of each step. It was found that CO2 reduction on the composite occurs most readily on the g-C3N4 side. The reaction path on the composite is different from that on g-C3N4. The heterojunction reduces the maximum energy barrier for CO2 reduction from 1.48 to 1.22 eV, following the optimal reaction path. Consequently, the BP/g-C3N4 heterojunction is theoretically proven to be an excellent CO2 reduction photocatalyst.This work is helpful for understanding the effect of BP modification on the photocatalytic activity of g-C3N4. It also provides a theoretical basis for the design of other high-performance CO2 reduction photocatalysts.
Key Words: Photocatalysis; CO2 reduction; Step-scheme heterojunction; Graphitic carbon nitride;Density functional theory
1 Introduction
Excessive CO2emission resulted from the combustion of fossil fuels leads to serious greenhouse effect and climate change1–3. It is urgent to explore clean and effective methods to reduce CO2release. Under this circumstance, photocatalytic CO2reduction arouses wide attention, because it converts harmful CO2into valuable hydrocarbons4,5. CO2is reduced assisted by photocatalysts and light irradiation6. Graphitic carbon nitride (g-C3N4) is a popular metal-free photocatalyst used for CO2reduction7,8. It has several advantages such as nontoxicity,narrow bandgap and good physicochemical stability9,10.Moreover, its considerably negative conduction band meets the thermodynamic requirement of CO2reduction to all kinds of products. However, the photocatalytic CO2reduction activity of pristine g-C3N4is extremely poor11.
Various strategies have been developed to enhance the photocatalytic activities of g-C3N4, among which the hybridization with others proves to be effective12–15. Enormous composites have been explored including CdMoO4/g-C3N416,BiVO4/g-C3N417, Bi2MoO6/g-C3N418, CdxZnyS/g-C3N419,graphene/g-C3N420, and Ti3C2/g-C3N421. Recently, black phosphorus (BP) has been employed to improve the photocatalytic performance of g-C3N4in diverse areas22,23. For example, Qiuet al.prepared BP decorated g-C3N4nanosheet through a post calcination process24. The optimal BP/g-C3N4composite exhibited a high nitrogen fixation rate of 347.5 μmol·L−1·h−1, exceeding Pt deposited g-C3N4. Konget al.synthesized BP quantum dot loaded g-C3N4photocatalystviaa high-vacuum stirring method25. With the BP percentage of 7%,the H2production rate was 3.6 times larger than that of unloaded g-C3N4. Wanget al.fabricated a BP/g-C3N4nanohybrid composed of BP quantum dot and tubular g-C3N426. The novel 0D/1D structure showed excellent photocatalytic activity towards Cr (VI) reduction and oxytetracycline hydrochloride degradation.
Nevertheless, report on CO2reduction of BP/g-C3N4photocatalyst is scarce. Hanet al.prepared BP quantum dot modified g-C3N4photocatalyst using an electrostatic attraction method and tested its CO2reduction activity27. The CO production rate of BP/g-C3N4was 2.5 times higher than that pristine g-C3N4. Although enhanced photocatalytic activities were achieved in the above examples, they merely speculated that promoted separation of photogenerated charge carriers were the intrinsic mechanism for the activity enhancement, while the essential function of BP modification was not explicitly investigated. There is still controversy over the role of BP. Some regarded it as a hole-migration cocatalyst whereas some deemed it as an electron mediator28,29. Others also claimed that BP/g-C3N4composite was a type-II heterojunction photocatalyst30.Besides, no study has paid attention to the CO2reduction reaction process on BP/g-C3N4composite.
Herein, to reveal the underlying mechanism of the improved CO2reduction performance of BP/g-C3N4composite photocatalyst, theoretical study using density functional theory calculations is conducted. A 2D/2D BP/g-C3N4composite model is built, and its photocatalytic behavior is meticulously evaluated. The geometric structure, electronic property, charge transfer, and CO2reduction reaction process of this model are systematically investigated. It is anticipated that this work can provide references for the interpretation of other g-C3N4-based or BP-modified photocatalysts.
2 Calculation methods
All the density functional theory calculations were carried out by using the Viennaab initiosimulation package31,32. The interaction between electrons and ion nucleus was implemented by the projector augmented wave (PAW) method33. The exchange-correlation interactions were performed utilizing the generalized gradient approximation (GGA) in the form of Perdew-Burke-Ernzerhof (PBE)34. DFT-D method of Grimme was employed to describe the van der Waals (vdW)interaction35,36. A 450 eV cutoff energy was adopted to expand the electron wave functions. For Brillion zone, the Monkhorst–Pack grid with a 3 × 3 × 1k-point was used for geometry optimization and property calculation37. A vacuum space of 1.5 nm was applied to eliminate interactions between neighboring images. In geometry optimization, all atoms were fully relaxed,and the convergence criteria for energy and force were set to 10−5eV and 0.1 eV·nm−1, respectively. Due to the well-known underestimation of band gap (Eg) in the GGA-PBE functional,the Heyd-Scuseria-Ernzerhof 2006 (HSE06) hybrid functional was adopted to acquire precise energy gap values38.
The change in Gibbs free energy (ΔG)39,40is defined as:
ΔG= ΔE+ ΔEZPE−TΔS+ ΔGpH+ ΔGU(1)where ΔEis the electronic energy difference, ΔEZPEand ΔSare the change in zero-point energy and entropy, respectively.Tis the temperature (298.15 K). ΔGPHis the correction of the H+free energy by the concentration, and it is equal tokB×T× ln10 ×pH, wherekBis the Boltzmann constant and pH is set to be zero for acidic condition. ΔGUis calculated by −neU, wherenis the number of transferred electrons andUis the electrode potential.Zero-point energies are calculated from the vibrational frequencies through fixing the catalyst on the bottom (the vibrations of substrate were ignored). The entropies of isolated gas are obtained from the NIST database. The adsorption energy(Eads) is computed by the formula:

whereEsub+a,EsubandEarepresent the total energies of substrate with adsorbed species, pure substrate without adsorbate, and pure adsorbate, respectively.
3 Results and discussions
3.1 Geometric structures
The optimized cell parameters of g-C3N4area=b= 0.71 nm,γ= 120°, and those of BP area= 0.45 nm,b= 0.33 nm,γ= 90°.These parameters are consistent with the values in previous experimental and theoretical studies41,42. 2D/2D BP/g-C3N4heterojunction was built by placing monolayer g-C3N4above the surface of monolayer BP. To realize a small lattice mismatch between the two components, a 2 × 2 monolayer g-C3N4supercell composed of 24 C atoms and 32 N atoms was constructed (Fig. 1a), and its lattice constant wasa=b= 1.42 nm. Meanwhile, rhombic monolayer BP containing 48 P atoms was cleaved from 2 × 3 bulk BP with the diagonal as the surface edge (Fig. 1b), and its lattice constant wasa=b= 1.48 nm.Subsequently, an appropriate BP/g-C3N4composite model was obtained by stacking the two layers.
The optimized structure of BP/g-C3N4heterojunction is shown in Fig. 1c. The g-C3N4layer in the composite presents a corrugated structure instead of the planar structure in pristine g-C3N4, and the distortion degree (h1) is 0.15 nm. The upright height (h2) of BP layer in the composite is 0.26 nm, larger than that in pristine BP (0.21 nm). The equilibrium distance (d)between BP and g-C3N4is 0.26 nm, in agreement with other g-C3N4-based heterojunctions43. The equilibrium distance conforms to the structural characteristics of vdW heterojunction,indicating that a vdW interaction is established between BP and g-C3N4. To examine the stability of the BP/g-C3N4composite structure, interface adhesion energy (Eadh) is evaluated by the following equation:
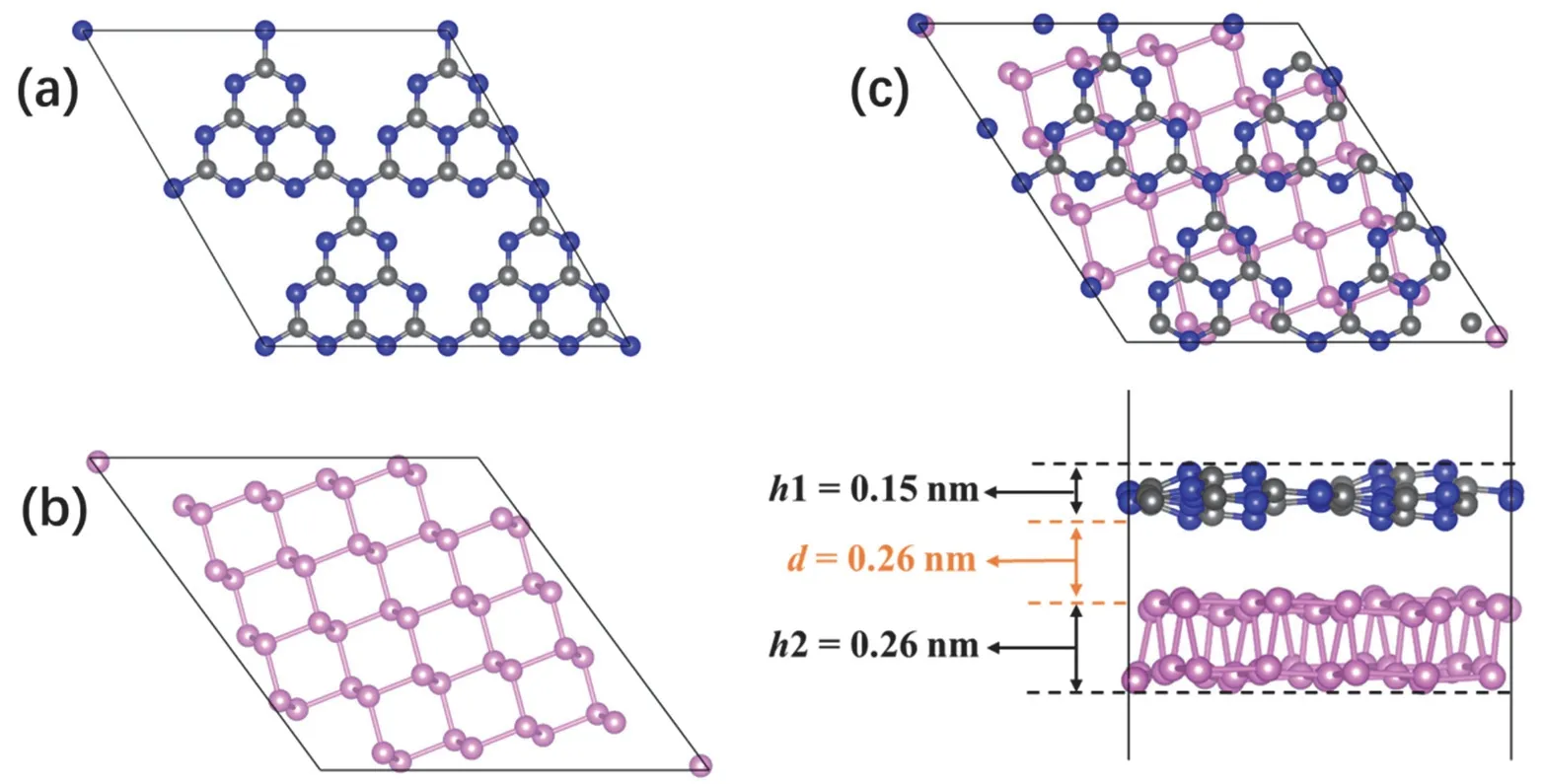
Fig. 1 Optimized geometric structures. Top views of (a) g-C3N4 and (b) BP. (c) Top view (upper) and side view (lower) of BP/g-C3N4 heterojunction.

whereE(BP/g-C3N4),E(g-C3N4) andE(BP) are the total energies of BP/g-C3N4hybrid, monolayer g-C3N4and monolayer BP,respectively. The calculatedEadhis −0.50 eV, indicating the formation of BP/g-C3N4is an exothermic process, suggesting the composite is thermodynamically stable.
3.2 Electronic properties
To reveal the photocatalytic mechanism in BP/g-C3N4heterojunction, electronic properties were investigated,including the density of states (DOS) and work function of BP and g-C3N4, and the charge density difference of BP/g-C3N4composite. The DOS was calculated using HSE06 hybrid functional. As shown in Fig. 2, the calculated band gaps of monolayer g-C3N4and BP are 2.78 and 1.48 eV, respectively, in consistency with the previous experimental and theoretical values44,45. The valence band (VB) of g-C3N4mainly consists of N 2porbital, and the conduction band (CB) of g-C3N4is composed of C 2pand N 2porbitals46. For BP, both the VB and CB are mainly dominated by P 2porbital.
Work function (Φ) is an important criterion for evaluating electron transfer. It is obtained by calculating the electrostatic potential alongZaxis direction and defined as:

whereEVacandEFrepresent the electrostatic potentials of vacuum level and Fermi level, respectively. As illustrated in Fig.3, the computed respective work functions of g-C3N4and BP are 4.28 and 4.54 eV, suggesting that the Fermi level of g-C3N4is higher than that of BP42,47,48. Therefore, it can be deduced that as the two layers incorporate, the Fermi level difference drives electrons transfer from g-C3N4to BP until an equilibrium Fermi level is achieved.
The electron transfer between the two components can be intuitively reflected by charge density difference (Δρ), which is calculated based on the following equation:

whereρ(BP/g-C3N4),ρ(BP), andρ(g-C3N4) are the charge densities of BP/g-C3N4heterojunction, monolayer BP and monolayer g-C3N4, respectively. Fig. 4a,b illustrate the threedimensional and planar-averaged charge density difference of BP/g-C3N4heterojunction, respectively. In three-dimensional view, the yellow and blue regions represent charge accumulation and depletion, respectively. In planar-averaged curve, the positive and negative Δρvalues signify charge density increase and decrease, respectively. In Fig. 4, it is clear that the yellow and blue regions are principally distributed in the interlayer space between BP and g-C3N4. As such, the main peak location in planar-averaged curve also provides the same information.These results sufficiently disclose the strong interfacial interaction in BP/g-C3N4heterojunction. Further observation shows that the yellow region is mainly distributed on the surface of BP, and the upper layer of g-C3N4is covered by blue region.Correspondingly, in the planar-averaged curve, the Δρvalue is primarily positive and negative at the BP and g-C3N4side,respectively. These finding convincingly demonstrates that the electrons in g-C3N4transfer to BP, in line with work function analysis. The interfacial electron transfer induces the formation of an interfacial built-in electric field, which is conducive to the later migration of photogenerated charge carriers.

Fig. 2 Calculated DOS of monolayer (a) g-C3N4 and (b) BP using HSE06 functional.

Fig. 3 Electrostatic potentials of monolayer (a) g-C3N4 and (b) BP.

Fig. 4 (a) Three-dimensional and (b) planar-averaged charge density difference of BP/g-C3N4 heterojunction.
Optical absorption, an important factor, is evaluated by calculating the absorption coefficient using the following formula:
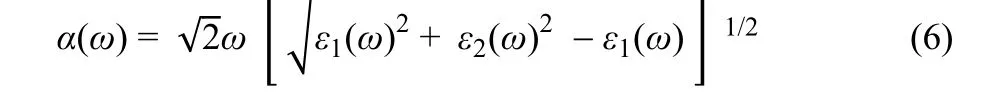
whereε1andε2represent the real and imaginary parts of the dielectric function, respectively;ωdenotes the light frequency.The result is depicted in Fig. 5. In a wide photo energy range of 1.5–4.0 eV, the absorption coefficient follows the order of BP/g-C3N4> BP > g-C3N4. Therefore, the hybridization of BP and g-C3N4results in increased light absorption, which is conducive to the photoexcitation of electrons and holes.

Fig. 5 Optical absorption coefficient of g-C3N4, BP, and BP/g-C3N4 composite.

Fig. 6 The S-scheme photocatalytic mechanism in BP/g-C3N4 heterojunction.
3.3 Photocatalytic mechanism
Based on the above electronic properties, the photocatalytic mechanism in BP/g-C3N4heterojunction is discussed. Initially,the band edge potentials are calculated using the following equations:

whereEVBandECBare the potentials of valence band and conduction band, respectively.χis the absolute electronegativity of the semiconductor, and it is 4.34 eV and 5.62 eV for g-C3N4and BP, respectively49,50.Eeis the energy of free electrons on the hydrogen scale (4.5 eV).Egvalues of g-C3N4and BP are 2.78 and 1.48 eV, respectively. Therefore, the obtained VB and CB potentials of g-C3N4are 1.23 and −1.55 eV, respectively, and those of BP are 1.86 and 0.38 eV, respectively. The calculated band edge potentials of g-C3N4are higher than those of BP,which is consistent with experimental results51,52. Judging from the results, a staggered band structure is constituted in BP/g-C3N4heterojunction.
Subsequently, the photocatalytic mechanism in BP/g-C3N4heterojunction is proposed and displayed in Fig. 6. After g-C3N4comes into contact with BP, electrons migrate from g-C3N4to BP because of its smaller work function and higher Fermi level. As a result, a built-in electric field pointing from g-C3N4to BP is formed. Meanwhile, as varying degrees of the electronic potentials at different regions, the CB and VB of g-C3N4bend up, and those of BP bend down. Under visible-light irradiation,both g-C3N4and BP are excited to generate electron–hole pairs.Driven by the collective effect of built-in electric field, band bending, and Coulomb attraction, photogenerated electrons in the CB of BP will combine with the photogenerated holes in the VB of g-C3N4. Accordingly, the photogenerated electrons with strong reduction ability in the CB of g-C3N4survive from recombination and energetically participate in CO2reduction reaction. Meanwhile, the photogenerated holes in the VB of BP have strong oxidation ability to induce oxidation reaction such as pollutant degradation. The above migration process of charge carriers conforms to the typical S-scheme photocatalytic mechanism53,54. In a word, BP/g-C3N4composite is a S-scheme heterojunction photocatalyst with both efficient charge separation and enough reduction capacity for CO2conversion.
3.4 CO2 reduction reaction process
Typically, CO2is converted into a series of products (such as CO and CH3OH)viasequential hydrogenation steps aided by photogenerated electrons55,56. In each step, H atom is added either on C or on O atom, thus resulting in various intermediates.According to the generation of different intermediates, some reaction paths are proposed and illustrated in Fig. 7. In general,the change in Gibbs free energy of each step is calculated and compared to determine the preferential reaction path and maximum energy barrier.
As for the reduction process from CO2to CH3OH on g-C3N4surface, an established reaction path has been acknowledged in many theoretical researches57,58, namely, CO2→ COOH* →CO → HCO* → H2CO → H3CO* → CH3OH. In view of the fact, we also chose this path to investigate the CO2reduction on g-C3N4surface, without considering other reaction paths. Fig. 8a shows the variation of Gibbs free energy in the CO2reduction process on g-C3N4surface. As can be seen, three steps with uphill energy are CO2→ COOH*, CO → HCO*, and H2CO →H3CO*, require energy input of 1.48, 0.19, and 1.12 eV,respectively. By contrast, the other three steps are downhill processes. Hence, the hydrogenation of CO2to COOH* with the largest ΔGis the rate-determining step, and the corresponding energy barrier is 1.48 eV. Previous reports also concluded that the hydrogenation of CO2is the most difficult step in the CO2reduction process on many g-C3N4-based photocatalytic systems58,59. Fig. 8b shows the optimized geometric structures of the reaction system. It is observed that COOH, HCO, and H3CO groups are linked with g-C3N4viathe formation of C―N or O―C bond, while CO2, CO, H2CO, and CH3OH molecules are adsorbed on g-C3N4.
Afterwards, the CO2reduction process on BP/g-C3N4heterojunction is explored. Prior to the exploration, it should be determined whether CO2reduction proceeds on the surface of g-C3N4or that of BP. On the basis of the aforementioned S-scheme photocatalytic mechanism, the photogenerated electrons in the CB of BP are consumed by the photogenerated holes in the VB of g-C3N4, while the separated photogenerated electrons in the CB of g-C3N4can trigger CO2conversion. In this regard, the CO2reduction reaction prefers to occur on the g-C3N4component in BP/g-C3N4composite. Besides, as CO2adsorption is influential for the later reduction process, the CO2adsorption on both side of BP/g-C3N4heterojunction is compared. Fig. 9 shows the optimized geometric structures. The calculated adsorption energy is −1.57 and −1.47 eV, respectively, manifesting the more stable adsorption on the g-C3N4side. Therefore, the CO2reduction on the g-C3N4side of BP/g-C3N4heterojunction is investigated.
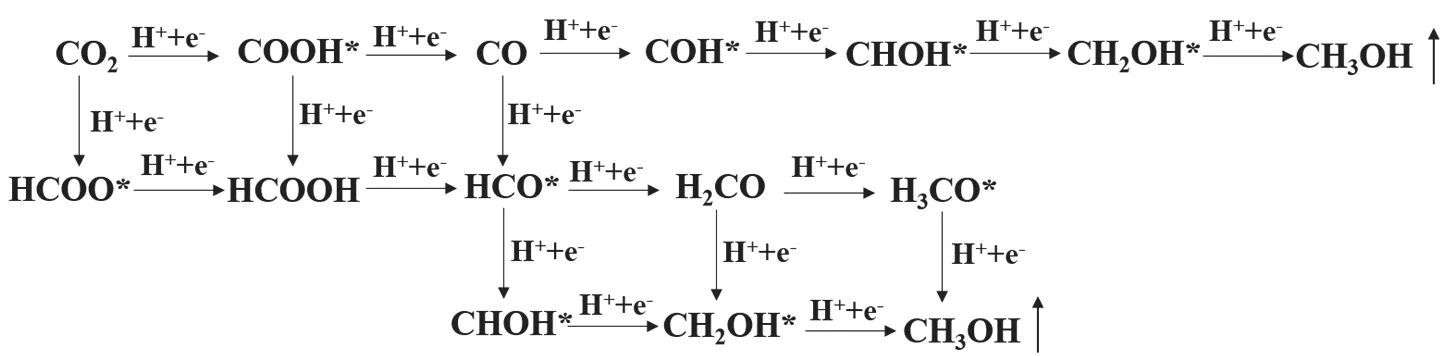
Fig. 7 Possible reaction paths of CO2 reduction to CH3OH.
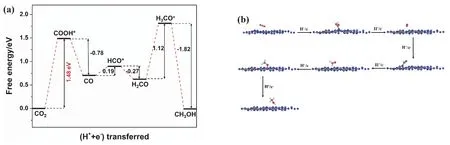
Fig. 8 (a) Free energy diagram of the reaction system and (b) optimized geometric structures in the CO2 reduction process on g-C3N4 surface.
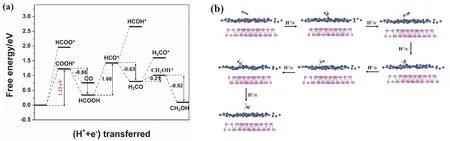
Fig. 10 (a) Free energy diagram of the reaction system and (b) optimized geometric structures in the CO2 reduction process on BP/g-C3N4 heterojunction.
Concerning the reduction process of CO2to CH3OH on BP/g-C3N4heterojunction, various reaction paths are examined. As shown in Fig. 10a, in the first hydrogenation step, the formation of COOH* requires a lower energy input of 1.22 eV than that of HCO O*, indicating that CO2is firstly reduced to COOH* rather than HCOO*. Then the COOH* is hydrogenated to HCOOH, or converted to COviadehydroxylation. The COOH* → HCOOH step releases energy of 0.88 eV, larger than the COOH* → CO step (0.47 eV). Therefore, HCOOH is a preferential intermediate product than CO. The adsorption energy of HCOOH is −1.92 eV.Subsequently, the HCOOH is further hydrogenated and dehydrated to produce HCO*, and the corresponding free energy is elevated by 1.08 eV. In the following process of HCO*hydrogenation, wide disparity exists between the reaction free energy of H2CO and CHOH*. The generation of H2CO is an exothermic process, while the production of CHOH* is an endothermic reaction. Hence, HCO* is more likely to be hydrogenated to H2CO. The adsorption energy of H2CO is −1.58 eV. The next-step hydrogenation of H2CO involves the generation of H3CO* and CH2OH*, in which the ΔGvalue is 0.80 and 0.21 eV, respectively. Thus, the final reduction step is continued by the hydrogenation of CH2OH* to CH3OH releasing 0.92 eV. The adsorption energy of CH3OH is −1.71 eV. Overall,the optimal reaction path of CO2reduction on BP/g-C3N4heterojunction is described as CO2→ COOH* → HCOOH →HCO* → H2CO → CH2OH* → CH3OH. Similarly, on pristine g-C3N4, the rate-determining step is also the hydrogenation of CO2to COOH* but with a decreased energy barrier from 1.48 to 1.22 eV. The lowered energy barrier indicates a more facile CO2reduction reaction process on BP/g-C3N4heterojunction.
Fig. 10b shows the optimized geometric structures of the reaction system corresponding to the optimized CO2reduction reaction path on BP/g-C3N4heterojunction. Analogous to the geometric structures on pristine g-C3N4surface, the COOH,HCO, and CH2OH groups are linked with the g-C3N4component in BP/g-C3N4compositeviathe formation of C–N bond, while the CO2, HCOOH, H2CO, and CH3OH molecules are adsorbed on the g-C3N4side of BP/g-C3N4composite.
4 Conclusions
In summary, geometric structures, electronic properties,photocatalytic mechanism, and CO2reduction reaction processes of 2D/2D BP/g-C3N4heterojunction are investigated by DFT calculations. Monolayer BP and g-C3N4constitute a stable BP/g-C3N4composite with negative adhesion energy of -0.50 eV and moderate equilibrium distance of 0.26 nm. Work function and charge density difference disclose the strong interfacial interaction. The electron transfer between BP and g-C3N4induces the formation of built-in electric field and band bending.As a result, the charge migration in BP/g-C3N4heterojunction follows S-scheme photocatalytic mechanism, and the photogenerated electrons with strong reduction ability are efficiently separated and accumulated in g-C3N4. Then the reaction process of CO2reduction to CH3OH on the g-C3N4side in BP/g-C3N4heterojunction is examined. The optimal reaction path is determined as CO2→ COOH* → HCOOH → HCO* →H2CO → CH2OH* → CH3OH. The hydrogenation of CO2to COOH* is the rate-determining step, and the corresponding energy barrier is 1.22 eV. By comparison, the traditional CO2reduction reaction path on pristine g-C3N4exhibits a maximum energy barrier of 1.48 eV. Overall, BP/g-C3N4S-scheme heterojunction is featured with efficient charge separation,strong reduction capacity, and facile CO2reduction reaction process; it is undoubtedly an outstanding CO2reduction photocatalyst.

- 物理化学学报的其它文章
- Fluorinated TiO2 Hollow Photocatalysts for Photocatalytic Applications
- Controllable Synthesis of g-C3N4 Inverse Opal Photocatalysts for Superior Hydrogen Evolution
- 微波辅助快速制备2D/1D ZnIn2S4/TiO2 S 型异质结及其光催化制氢性能
- TiO2-Supported Single-Atom Catalysts for Photocatalytic Reactions
- Carboxyl-Functionalized Graphene for Highly Efficient H2-Evolution Activity of TiO2 Photocatalyst
- Review of Z-Scheme Heterojunctions for Photocatalytic Energy Conversion