
功能化金属有机框架材料催化二氧化碳转化研究进展
2021-07-10 07:08:16廖再添陈治洲孙为银
无机化学学报 2021年7期
赵 丹 廖再添 张 旺 陈治洲 孙为银
(1宁波大学海洋学院,宁波 315832)
(2南京大学化学化工学院,配位化学国家重点实验室,南京 210023)
(3温州大学电气与电子工程学院,温州 325035)
0 引 言
金属有机框架(metal-organic frameworks,MOFs)是以金属离子或金属簇为节点与有机桥联配体通过配位作用形成的具有特定网络结构的功能性材料,是近年来在无机化学与晶体工程交叉领域研究最为活跃的领域之一[1-5]。MOFs 材料有比表面积大、孔径及孔道结构可调、金属及有机配体丰富多样、易于功能化等诸多优点,使其在气体吸附存储与分离、磁性材料、电子与质子传递、生物及化学传感、光电材料、分子识别与拆分、生物医学和多相催化等广阔领域具有潜在的应用价值[6-12]。近年来,MOFs 在催化领域的应用研究越来越广泛[13-14]。MOFs 结构中排列紧密、均匀分散的催化活性位点有利于加速催化反应;并且其较多的孔结构也进一步确保了每个催化活性位点与反应底物的接触;同时,MOFs 结构中较大的孔道有利于运输催化反应底物和产物。此外,由于MOFs 结构和功能独特的可设计性及可调性等特点,可以通过改变有机配体和金属中心的种类,进而实现具有高效催化性能的新型MOFs 材料的设计。由于MOFs 材料在催化方面的良好性能,目前以其作为非均相催化剂来捕捉并固定二氧化碳(CO2)已成为其应用的一个重要方向[15]。MOFs 材料的框架结构可以有效地捕捉CO2分子作为反应物,同时其结构中存在的金属配位不饱和位点和活性有机官能团可以作为反应的活性位点进一步催化CO2转化。在MOFs 的有机配体中引入活性基团,如路易斯碱、配位基团等,也可以提高MOFs 的催化活性。本文主要从功能化MOFs 催化剂的结构特性入手来介绍其在催化转化CO2方面的研究进展,并对该领域存在的问题和解决策略进行了总结,最后对其未来的发展趋势进行了展望。
1 功能化MOFs的催化作用模式
多孔MOFs 材料应用于催化具有反应空间效应(即MOFs 孔洞/孔道大小对反应底物/产物的选择性)、可循环使用等特点。另外,MOFs材料的可调性很强,它的孔径大小、形貌、晶面等可以通过金属中心和有机配体的选择和修饰以及合成条件的控制等方式得到精确的调控,研究者可以根据催化反应需求设计合成特定的MOFs,从而优化催化效果。MOFs 催化材料的作用模式主要有以下3 种[16-22]:(1)金属活性位点、(2) 活性功能基团、(3) 孔道效应(图1)。金属离子的配位不饱和位点可作为催化反应的活性中心;有机配体中存在的官能团可直接作为催化反应的活性中心,或者对其进行后修饰、改性后形成新的活性中心;MOFs 结构孔洞/孔道可作为催化反应的场所或载体,采用吸附、浸渍、沉淀等物理化学方法将活性位引入到MOFs 的孔道中,使反应在MOFs孔道内进行。
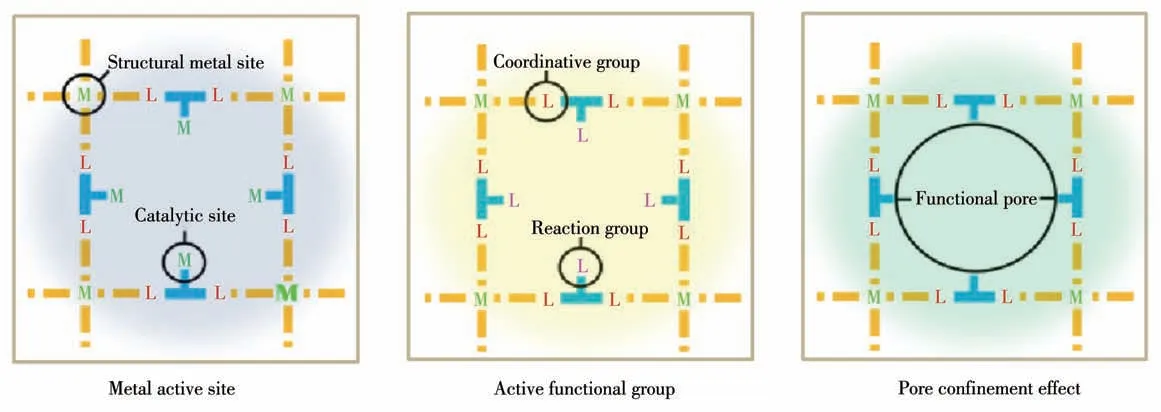
图1 MOFs的催化作用模式Fig.1 Catalytic modes of MOFs
2 功能化MOFs 对二氧化碳的催化转化
CO2作为碳基燃料产生的主要产物,近几十年一直是科学家关注的焦点,在CO2的转化利用方面也取得了诸多进展[23-25]。但是,相比于人类生产活动排放的CO2量级,转化利用的CO2量微乎其微。同时,CO2又是自然界中来源最为丰富和清洁的C1资源,如果直接将其化学转化则需要消耗大量的能量,因此实现CO2资源化有效利用的关键问题是如何实现它的高效转化[26]。
目前,活化CO2的方法主要包括:生物法、光化学还原、电化学还原、热还原以及金属配位活化等,也可使用以上的混合方法[25]。其中研究最为深入的是过渡金属配位活化CO2,其主要的活化方式有以下5种(图2):ガ过渡金属的p电子或d电子向CO2分子的反键轨道提供电子对,降低成键轨道能量,富电子的金属经常会采用这种方式与CO2配位发生M→C 的电子转移作用;ギCO2分子中一个氧原子的孤对电子与缺电子的金属中心配位发生O→M 作用;バCO2分子作为一个双齿配体通过2 个氧原子与缺电子的金属配位,氧原子与金属之间存在着O→M 的电子转移作用;ビCO2分子的电子与金属中心的空轨道形成σ键,同时金属中心的d轨道与CO2分子的空反键轨道形成反馈π键而发生电子转移;ブ金属中心与CO2分子中的C=O 双键作用形成一个侧面配位的π-配合物。总之,金属原子通过与CO2分子之间的作用,改变其分子中电子结构,从而实现对其活化。除此之外,有机官能团还可通过偶极-四极矩作用、氢键作用和孤对电子-π作用等非共价作用模式实现CO2分子的活化。
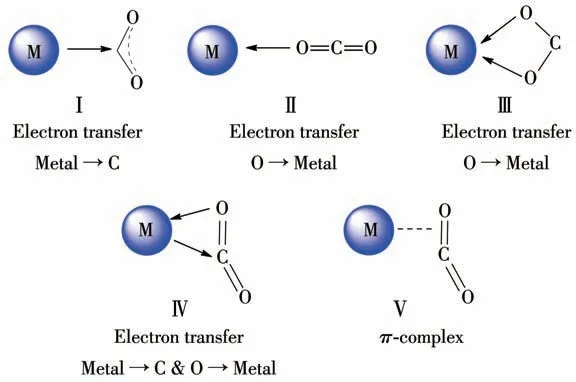
图2 金属中心活化CO2的方式Fig.2 Activation modes between CO2 and metal centers
实现CO2分子的转化不仅利于生态环境的保护、促进碳循环、缓解温室效应,更能降低生产成本、提高经济效益,因此,开发活化CO2分子的高效催化剂是最有潜力的解决途径[26]。近年来,经过科学家的不断努力,利用功能化MOFs 作为催化剂在CO2催化转化方面已经取得了可喜的成果。目前利用功能化MOFs 实现催化转化CO2的反应主要有2大类(图3):(1)CO2碳酸酯化反应、(2)CO2还原反应。下面主要介绍功能化MOFs 在这2 大类反应中近期的研究成果。
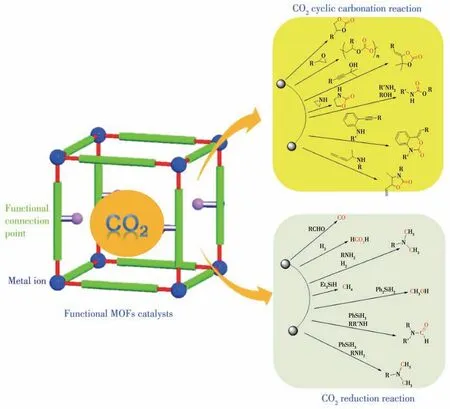
图3 功能化MOFs催化转化CO2的反应类型及其产物Fig.3 Reactions and products of CO2 conversion catalyzed by functional MOFs
2.1 功能化MOFs在CO2碳酸酯化反应中的应用
环状碳酸酯可作为惰性非质子极性溶剂及聚碳酸酯(常见的工程塑料成分)、医药和其他化学化工产品前驱体,是化工生产工艺中不可或缺的物质。利用催化剂将CO2转化为环状碳酸酯,是CO2化学转化的有效途径之一[29-31]。因为MOFs 材料在催化方面的良好性能,目前以其作为非均相催化剂来捕捉及化学固定CO2已成为其应用的一个重要方向[32-33],一些功能化MOFs 催化剂在CO2碳酸酯化反应中已表现出很大的潜力(表1)。下面将根据不同的催化活性中心介绍近几年功能化MOFs 在CO2环碳酸酯化反应方面的研究工作。
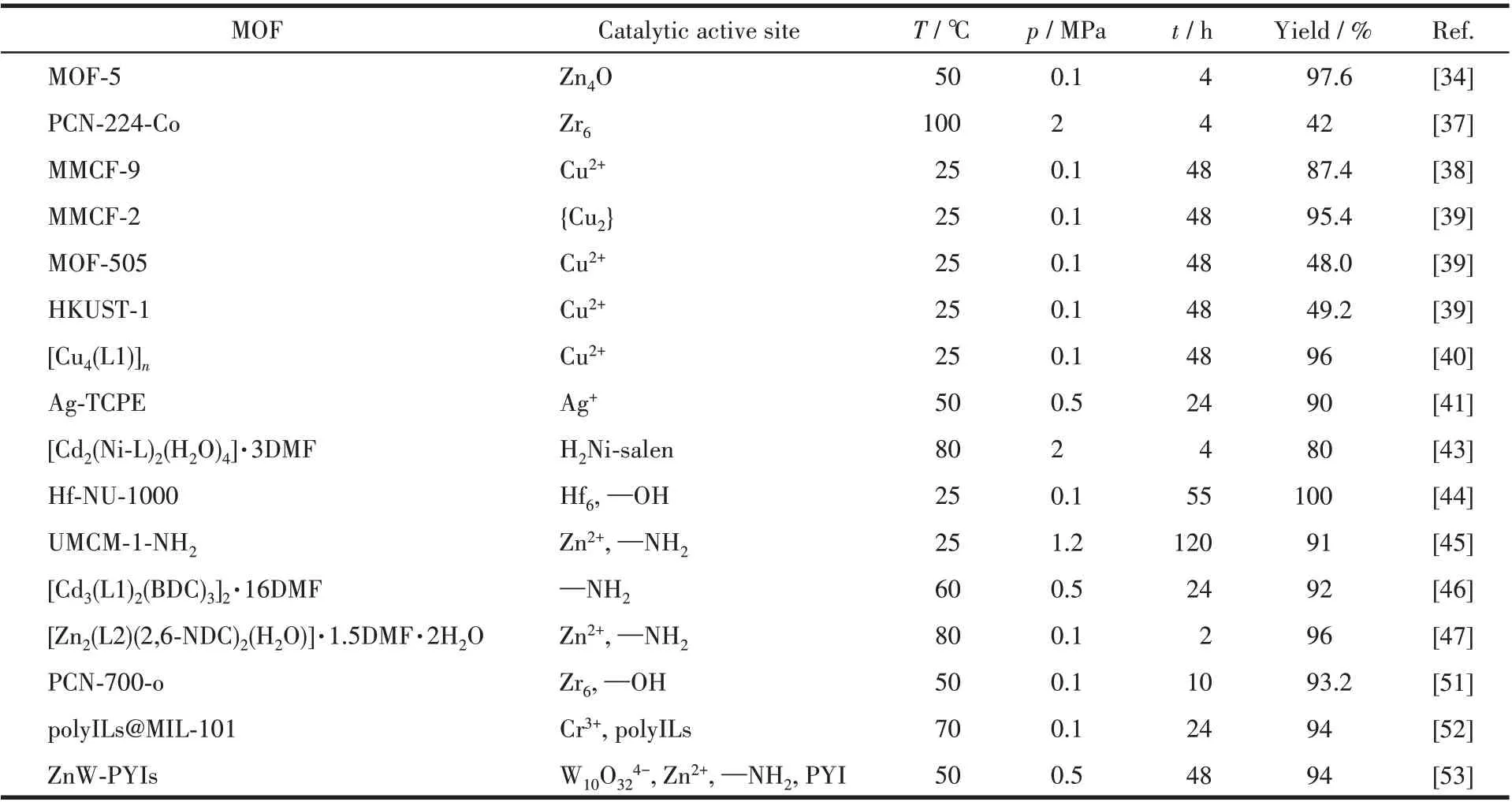
表1 一些功能化MOFs作为催化剂在CO2碳酸酯化反应中的性能Table 1 Catalytic performance of some reported functional MOFs catalysts in CO2 cyclic carbonation reaction
2.1.1 金属位点催化
MOFs 材料中金属中心以及配位不饱和金属离子(与金属离子配位的溶剂分子离去后产生)可以作为Lewis 酸性活性位点催化反应。在2009 年,Han等[34]就利用经典的MOF-5:[Zn4O(BDC)3](1)(BDC=对苯二甲酸)作为异相催化剂催化CO2的环加成反应,这也是首例基于MOFs 的多相催化环氧化合物与CO2的环加成反应(图4a)。作者系统地探究了影响反应的各项因素,并最终选择较为温和的反应条件(0.1 MPa CO2氛围、50 ℃)下反应4 h 后得到环碳酸酯,产率达97.6%,并且1 经分离回收重复使用3 次未见产率明显下降。催化机理表明1结构中的Zn4O簇不仅起连接骨架的作用,而且还作为Lewis 酸催化中心与环氧化合物连接并将其活化,作为共催化剂nBu4NBr中的Br-离子进攻被活化的环氧化合物使其开环并与CO2连接,实现整个催化循环过程(图4b)。除此之外,ZIF-8、Mg-MOF-74、MIL-125(Ti)、ZIF-68 以及MIL-101(Cr)等[35-36]都已被报道可作为异相催化剂用于CO2的环加成反应。

图4 MOF 1催化CO2环碳酸酯化反应示意图(a)和反应机理(b)[34]Fig.4 Schematic digram(a)and mechanism(b)for CO2 cyclocarbonation reaction catalyzed by MOF 1[34]
2013 年,Zhou 等[37]报道了以Zr6簇为节点与四(4-羧基苯基)卟吩(H2TCPP)在溶剂热条件下自组装得到了稳定PCN-224(无金属卟吩以及Ni-、Co-、Fe-卟啉),并利用PCN-224(Co) (2)结构中的Zr6簇作为Lewis 酸催化中心催化CO2和环氧丙烷的环加成反应(图5a和5b)。该系列化合物中均含有一个新型六连接的Zr6,孔隙率高达78.9%,BET 比表面积为2 600 m2·g-1,同时该系列化合物在pH=0~11 范围内能保持晶体稳定及气体吸附性能。催化实验结果表明在CO2压力为2 MPa、反应温度为100 ℃时,以nBu4NCl为助催化剂,反应4 h后产率可达到42%,TON 值达到461。而且催化剂经3 次循环使用后转化率没有明显降低。通过对催化剂进行X射线粉末衍射(PXRD)表征可知,催化剂2 的晶态结构保持完好(图5c)。
2014 年,Ma 课题组[38]报道了一例含金属Cu 的MOF 材料MMCF-9 (3),并且将其作为固相Lewis 酸催化剂高效催化CO2与环氧化合物反应生成相应的环碳酸酯。3 是由Cu(NO3)2·2.5H2O 和H10tdcbpp(四(3,5-二羧基联苯)卟吩)经溶剂热合成,加热处理后,结构中的部分Cu离子呈现出配位不饱和的Lewis酸活性位(图6a)。以nBu4NBr 为助催化剂,在常温常压下反应48 h 后,环氧丙烷的转化率达到87.4%,对反应后的催化剂进行回收利用,转化率仍可达到86.4%。同年,该课题组[39]以1,4,7,10-四氮杂环十二烷-N,N′,N″,N‴-四-对甲基苯甲酸(H4tactmb)为有机配体,与Cu(NO3)2·2.5H2O 反应构筑了分子式为[Cu2(Cu-tactmb)(H2O)3(NO3)2]的MOF:MMCF-2 (4)(图6b)。单晶结构分析表明,MOF 4 结构中6 个tactmb配体和12 个{Cu2}轮桨状次级结构单元(SBU)连接成一个立方八面体笼状空腔,每个{Cu2}上的Cu2+离子均为配位不饱和形式。基于MOF 4 结构中高密度的{Cu2} Lewis 酸活性位点,作者将其作为催化剂探究了其催化CO2与环氧化合物的环加成反应的催化活性(图6c)。研究结果表明,以nBu4NBr 为共催化剂,MOF 4 在常温常压下催化环氧丙烷与CO2的反应48 h 后的反应产率高达95.4%。在同样反应条件下,MOF-505 和HKUST-1 催化体系产率分别只有48.0%和49.2%。作者认为MOF 4 中Cu2(CO2)4结构连接整个框架使其整个结构中含有高密度的Lewis酸活性位点,因此对CO2的环加成反应显示出了优越的催化活性。可能的催化机理:{Cu2} Lewis 酸位点活化环氧化合物,Br-亲核进攻开环,CO2插入闭环,与MOF 1的催化机理一致(图6d)。
2016 年,Zhao 课题组[40]基于富氮的三氮唑配体H8L1 和铜盐反应合成了一例含有Lewis 酸金属位点的大孔MOF [Cu4(L1)]n(5)材料(图7a)。通过气体吸附和拉曼光谱证明MOF 5对CO2具有很强的亲和力(图7b)。该MOF 材料在常温条件下对CO2的高吸附能力以及Lewis 酸位点使其对CO2与环氧小分子反应生成环状碳酸酯反应具有高的催化活性:常温常压下反应48 h 产物产率可达到96%。当底物尺寸增加,催化活性减弱,说明CO2与环氧小分子反应生成环状碳酸酯是发生在该MOF 材料的孔道内(图7c)。
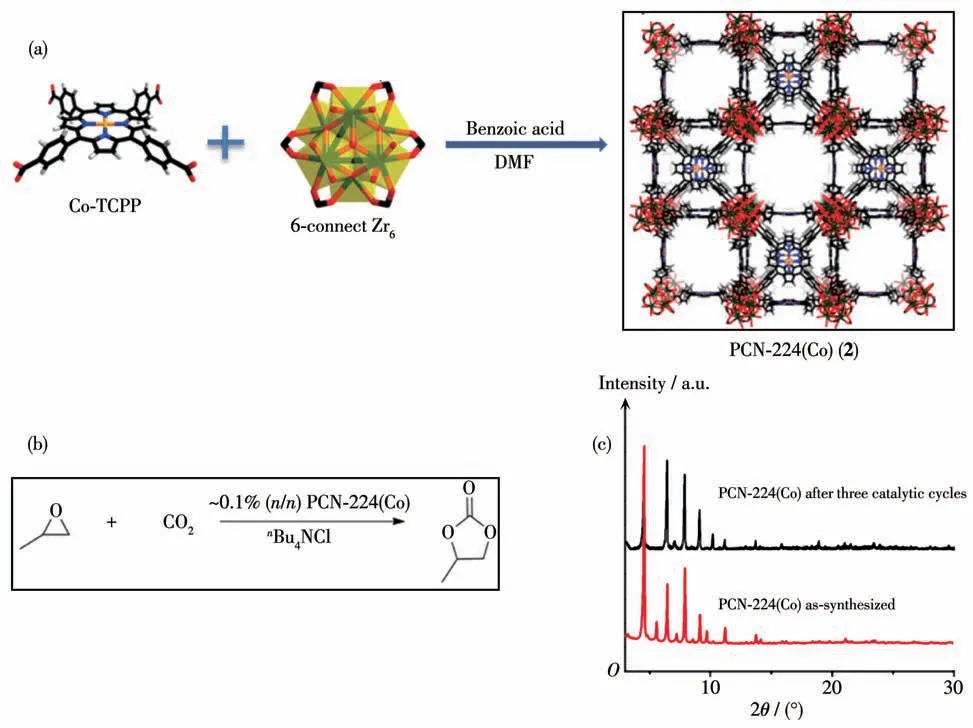
图5 (a)MOF 2的合成步骤和晶体结构;(b)MOF 2催化CO2和环氧丙烷的环加成反应;(c)MOF 2催化反应前后的PXRD图[37]Fig.5 (a)Synthetic route and crystal structure of MOF 2;(b)CO2 and propylene oxide coupling reaction catalyzed by MOF 2;(c)PXRD patterns of MOF 2 before and after three catalytic cycles[37]
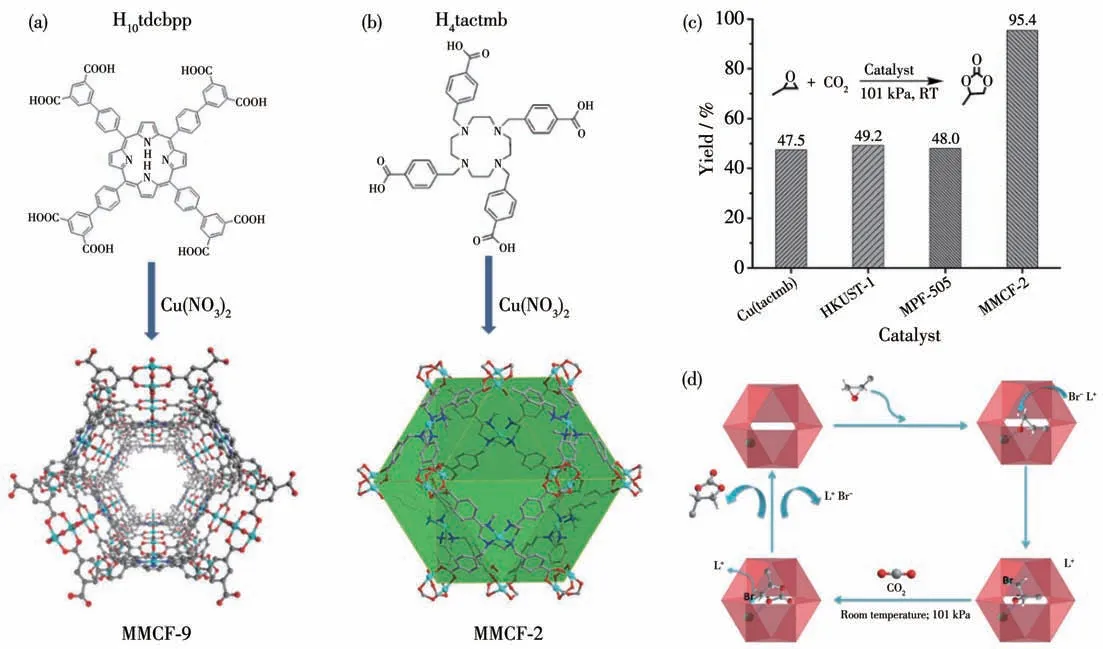
图6 (a)MOF 3的合成与结构;(b)MOF 4的合成与结构;(c)不同催化剂催化CO2和环氧丙烷反应的催化效率;(d)MOF 4催化CO2环碳酸酯化反应机理[38-39]Fig.6 (a)Synthesis and structure of MOF 3;(b)Synthesis and structure of MOF 4;(c)Catalytic efficiency of different catalysts for CO2 cycloaddition reaction;(d)Mechanism for CO2 cycloaddition reaction in the presence of MOF 4[38-39]
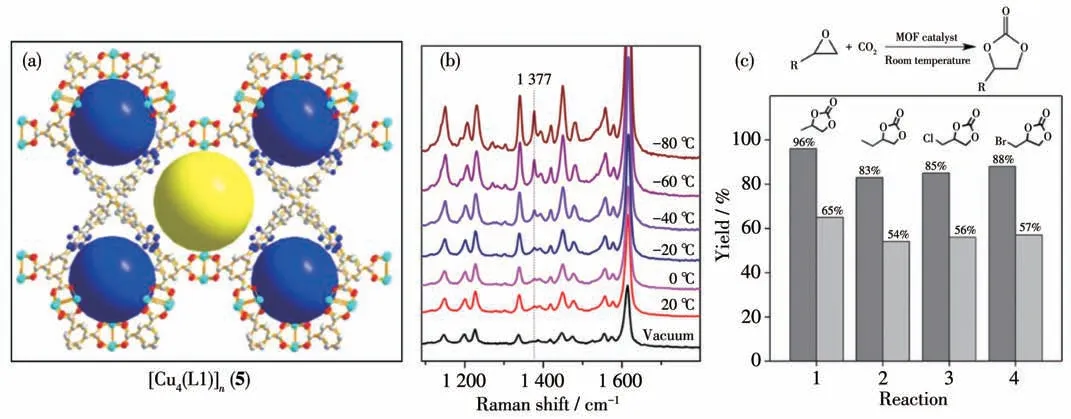
图7 (a)MOF 5的结构;(b)不同温度下MOF 5与CO2作用的拉曼光谱;(c)MOF 5(深色)和HKUST-1(浅色)催化的不同底物的CO2环加成反应[40]Fig.7 (a)Structures of MOF 5;(b)Raman spectra of MOF 5 bonding with CO2 at different temperatures;(c)CO2 cycloaddition reaction with different substrates catalyzed by MOF 5(black)and HKUST-1(gray)[40]
2017 年,Duan 课题组[41]报道了一例由四苯乙烯四羧酸(H4TCPE)为有机配体,一维Agガ链为次级结构单元构筑的三维框架网络结构Ag-TCPE (6)。考虑到MOF 6 结构中的一维Agガ链可以利用π-π相互作用来活化C≡C 键,因此他们探究了MOF 6 作为催化剂催化CO2与丙炔醇的环化反应(图8a)。催化实验结果表明在CO2压力0.5 MPa、反应温度50 ℃、乙腈为溶剂、三苯基膦为有机碱的条件下大部分底物的转化率都可达到90%以上。Duan 等捕捉到了在同一单晶中丙炔醇被俘获和活化的2种状态,直接展示了Ag+对炔基的π-活化作用,对苯乙炔的叠氮化反应也证实了Ag+的催化作用(图8b)。不仅如此,他们还进一步探索了催化串联反应的可能性。考虑到Ag+的σ-活化与π-活化作用,他们以苯乙炔为催化反应底物,将丙酮、叔丁醇钾、三苯基膦、乙腈、MOF 6 催化剂在CO2压力0.5 MPa、反应温度40 ℃条件下利用两步一锅法获得产率为93%的α-烷叉环碳酸酯类产物。
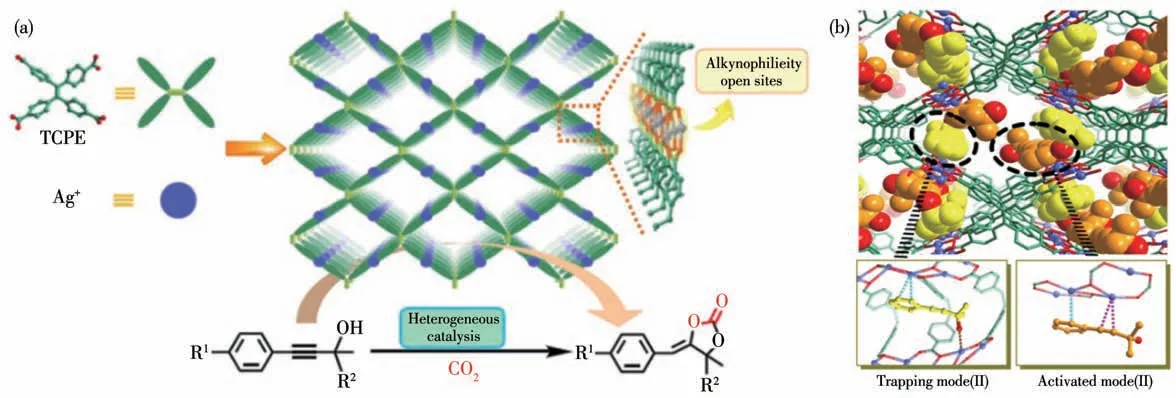
图8 (a)MOF 6的合成过程及其催化苯基丙炔醇与CO2环化形成环碳酸酯示意图;(b)MOF 6与苯基丙炔醇的作用[41]Fig.8 (a)Synthetic process of MOF 6 and representation of using MOF 6 as catalyst for the carboxylic cyclization of phenyl propargylic alcohol with CO2 to cyclic carbonates;(b)Interactions between phenyl propargylic alcohol and MOF 6[41]
2.1.2 金属位点和有机官能团协同催化
在MOFs 材料中,配体上的官能团如磺酸基、磷酸基、氨基、酰胺基、咪唑基等可增强多孔材料对CO2的捕集效果[41]。配体对CO2分子的亲和能力越高,对CO2的反应分压就会越小,也更具实际应用潜力。通过对配体的预设计或后修饰可有效提高MOFs整体的CO2催化转化能力。
2013 年,Jiang 课题组[43]通过对配体的预设计将席夫碱(salen)与Ni(OAc)2·4H2O 进行预配位形成优势催化配体H2Ni-salen,并通过与Cd2+自组装得到了一例多孔MOF 并将其作为异相催化剂来实现CO2环加成反应。催化结果表明在CO2压力2 MPa、反应温度80 ℃条件下,四丁基溴化铵的质量分数为3%时,反应4 h 后环碳酸酯的产率为80%。以配体或金属盐替换MOF 均不能达到很好的催化效果,证明了MOFs 框架组分对转化的重要作用。随后,Farha课题组[44]在溶剂热条件下合成了具有Hf6簇的MOF材料Hf-NU-1000 (7)(图9a)。在常温常压下,MOF 7催化氧化苯乙烯与CO2的环加成反应55 h后的产率可以达到100%,而当温度提高到55 ℃时反应在12 h 之内便可以进行完全(图9b)。作者认为Hf6簇上的—OH 具有较高的Brønsted 酸性是该催化剂高效的主要原因(图9c)。
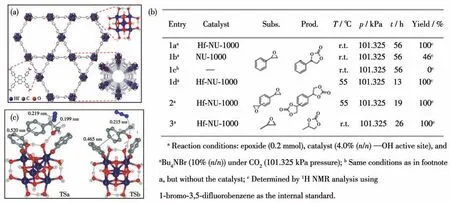
图9 (a)MOF 7的结构;(b)MOF 7催化CO2与不同底物的环加成反应;(c)MOF 7催化CO2环加成反应的催化机理[44]Fig.9 (a)Structure of MOF 7;(b)Catalytic efficiency of MOF 7 for CO2 cycloaddition reaction with different substrates;(b)Possible catalytic reaction pathway for CO2 cycloaddition reaction in the presence of MOF 7[44]
Park 等[45]利用2 种羧酸配体H3BTB(1,3,5-三(4-羧基苯基)苯)和H2BDC-NH2(2-氨基对苯二甲酸)在溶剂热条件下和锌盐反应得到了命名为UMCM-1-NH2的MOF 8,并利用其作为催化剂实现CO2的环加成反应(图10a)。实验结果表明在CO2压力为1.2 MPa、nBu4NBr 质量分数为0.64%的条件下,室温反应24 h时反应产率不到3%,而当温度为120 ℃时,其产率却达到了91%。当反应温度在室温至50 ℃区间时,其TOF 随温度的提高而增大(图10b)。反应后,催化剂可通过简单过滤分离出来。在数次循环使用后,催化剂结构未发生改变且产率没有明显的下降。催化机理实验结果表明MOF 8 结构中的Zn 离子可以作为Lewis 酸活性位点活化环氧化合物,—NH2活性位点不但可以增强CO2的吸附能力还可以作为Lewis 碱活化CO2,nBu4NBr 解离出来的Br-亲核进攻环氧底物使其开环,活化后的CO2插入后闭环完成整个环加成反应(图10c)。
近年来,本课题组构筑了一系列功能化MOFs并利用其作为催化剂实现CO2的碳酸酯化反应。在2017 年,我们设计合成了一例氨基三脚架咪唑配体N1-(4-(1-咪唑基)苄基)-N1-(2-氨乙基)乙二胺(L1)[46],并利用该配体与H2BDC 及镉盐在溶剂热条件下反应得到多孔动态功能化MOF 材料[Cd3(L1)2(BDC)3]2·16DMF (9)。单晶结构分析表明MOF 9 沿b轴方向上有较大孔道的三维五重互穿结构,其框架中—NH2官能团有序地排列在孔道表面。除此之外,MOF 9 具有独特的动态结构,其五重互穿结构间的相对位置移动使其对催化反应底物具有选择性(图11)。活化去除溶剂后,对该功能材料的吸附性能和催化CO2环加成反应进行研究发现:—NH2活性位点不仅有利于CO2的吸附,而且对CO2和丙炔胺的环化反应显示出优良的催化活性(TON 值可以达到9 300)。
随后,我们又报道了一例含有氨基三脚架三氮唑配体N1-(4-(1-1,2,4三氮唑)苄基)-N1-(2-氨乙基)乙二胺(L2)和2,6-萘二羧酸(2,6-H2NDC)的多孔MOF:[Zn2(L2)(2,6-NDC)2(H2O)]·1.5DMF·2H2O (10)[47]。结构分析结果表明MOF 10 中含有一维孔道结构,而且—NH2活性位点均匀分布在一维孔道中。活化去除溶剂分子后得到MOF 10′。MOF 10′结构中的配位不饱和位点Zn 可以作为Lewis 酸活性位,在常温常压条件下可以实现CO2的环加成反应(图12)。催化实验结果表明在常压条件下,MOF 10′对CO2与环氧化合物的环加成反应表现出了较高催化活性,其催化CO2和环氧底物的TOF 值达2 450 h-1。催化机理实验表明配体上的—NH2活性位点不但可以增强CO2的吸附能力还可以作为Lewis 碱活化CO2,与Lewis 酸活性位Zn 协同作用来实现CO2的环加成反应。并且MOF 10′的框架结构表现出对催化底物分子尺寸的选择性。
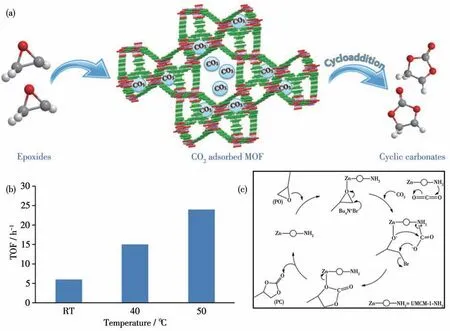
图10 (a)MOF 8的结构及其催化CO2环加成反应示意图;(b)不同温度下MOF 8作为催化剂催化CO2环加成反应的TOF值;(c)MOF 8催化CO2环加成反应的催化机理[45]Fig.10 (a)Schematic digram of MOF 8 for CO2 cycloaddition reaction;(b)TOF values of CO2 cycloaddition reaction using MOF 8 as catalyst under different temperatures;(b)Catalytic mechanism for CO2 cycloaddition reaction in the presence of MOF 8[45]
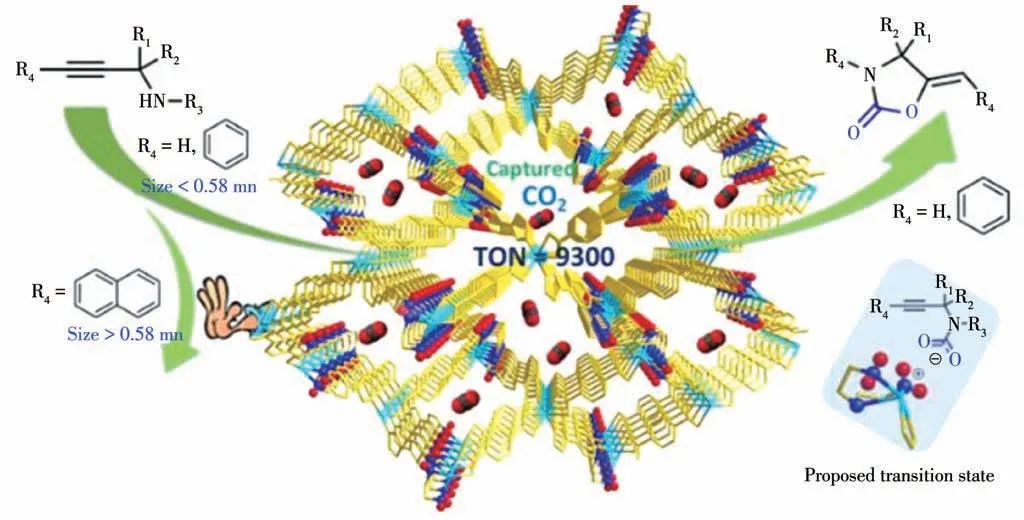
图11 MOF 9的结构及其催化CO2环加成反应示意图[46]Fig.11 Structure and schematic digram of MOF 9 for CO2 cycloaddition reaction[46]
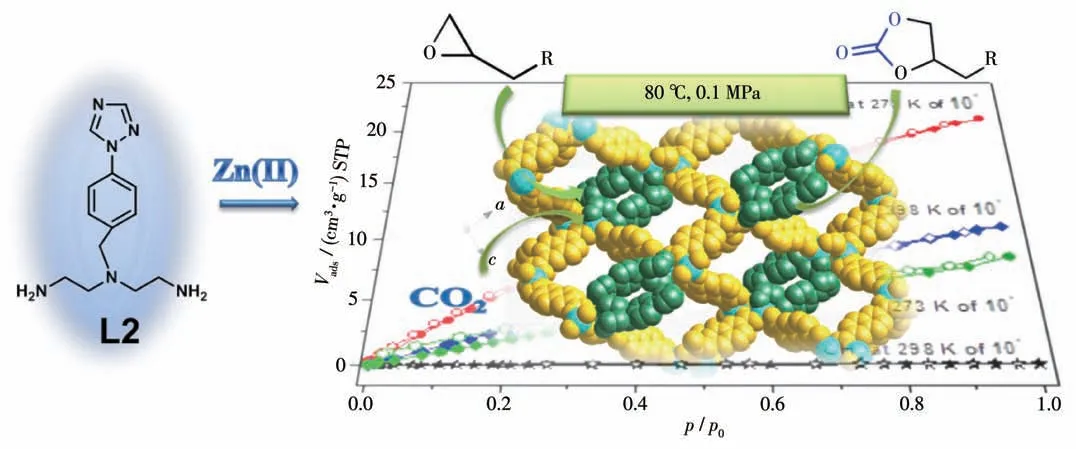
图12 MOF 10′的合成路线、结构及其吸附催化CO2环加成反应示意图[47]Fig.12 Synthetic process,structure and schematic digram of MOF 10′for CO2 cycloaddition reaction[47]
2.1.3 空腔负载和限域催化
MOFs 不仅本身是优良的多相催化剂,MOFs 结构的多孔性还使其可以作为催化剂的载体。同时,MOFs 孔道的限域效应能很好地实现对底物分子的选择性催化,为后续的产物分离提纯提供了便利。对于环加成反应来说,MOFs 骨架上的金属不饱和配位点仍作为Lewis 酸活性中心,与负载物起到协同作用降低反应能垒,孔道尺寸起到对底物的筛选作用。最常作为载体的MOFs 框架有ZIF-8、UIO-66、UIO-67、MIL-101(Cr、Fe)等[48-50]。
2016 年,Zhou 等报道了具有呼吸效应的含有Zr6簇结构的MOFs PCN-700 系列[51]。该系列MOFs采用不同数量甲基或三氟甲基修饰的联苯二羧酸配体与Zr6金属节点构筑框架。PCN-700 系列MOFs的空腔开合来源于配体2 个苯环之间C—C 键的扭转,从而导致Zr6簇与配位羧基角度的改变,进而实现框架整体的开合程度变化。结构分析表明PCN-700-o (11)框架沿c轴方向的长度为1.492 nm,而去除溶剂分子后得到PCN-700-c(11′),沿c轴方向的长度变为1.124 nm。MOF 11′在使用DMF 或三氟乙酸处理后孔道又可恢复为1.492 nm,实现了孔道开合的可控性(图13a)。在催化CO2环加成实验中,开放型框架MOF 11 具有很好的催化效率及底物扩展性,而闭合型框架MOF 11′的催化活性则大幅下降(图13b)。
2018 年,Jiang 课题组[52]基于MIL-101 后修饰制备了一例负载聚合物催化剂的MOF 并命名为polyILs@MIL-101(12)。该材料是在MIL-101 中同时加入1-乙烯基-3-乙基咪唑溴化盐(VEIMBr)和邻二乙烯基苯(o-DVB)原位合成聚咪唑离子液体(polyILs),实现咪唑溴化盐的固载,成功制备了同时包含Lewis 酸和Lewis 碱催化位点的复合材料(图14a)。在催化CO2与环氧乙烷衍生物的反应中,由于咪唑溴化盐可提供反应所需的Br-,故无需额外添加共催化剂。咪唑溴化盐的负载不仅增加了MOF 12 对CO2气体的捕获能力,使复合材料能在较低的CO2气压下实现高效转化;同时可以不外加四丁基溴化铵等亲核试剂实现了亲核催化组分的多相化,降低了分离回收成本。实验表明随CO2分压的降低,MOF 12 中各催化组分的协同作用随之凸显,远高于咪唑溴化盐的催化转化率(图14b)。该复合材料可对多种环氧化物与CO2的反应进行催化,并在10次循环再利用后依然保持高的催化活性(图14c)。
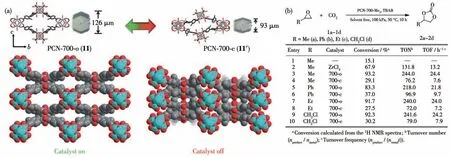
图13 (a)MOFs 11与11′的结构转变过程;(b)MOFs 11和11′对CO2环加成反应的催化效率[51]Fig.13 Structural transformation between MOFs 11 and 11′;(b)Catalytic efficiency of MOFs 11 and 11′for CO2 cyclization reaction[51]
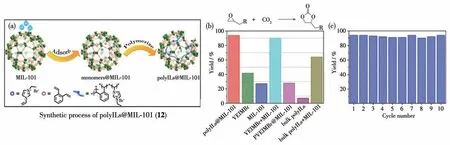
图14 (a)MOF 12的制备过程示意图;(b)不同催化剂对CO2环加成反应的催化效率;(c)MOF 12催化剂的循环实验结果[52]Fig.14 (a)Schematic illustration showing preparation of MOF 12;(b)Catalytic efficiency of different catalysts for CO2 cyclization reaction;(c)Histogram of recyclibility study for catalytic activities of MOF 12 for CO2 coupling reaction[52]
2.1.4 多组分协同催化
MOFs 孔道具有可修饰性,MOF 各组成部分可通过预合成、后修饰等方式实现多种功能的集中和协同,相比其他多相催化剂更具备多组分协同催化的潜力。Duan 课题组[53]利用Keggin 型多金属氧酸盐([ZnW12O40]6-)、Lewis 酸性金属(Zn2+)、氨基联吡啶(NH2-BPY)和手性有机配体2-吡咯烷基咪唑(PYI)设计构筑了包含多种催化能力的MOF ZnW-PYIs(13),并实现了低碳烯烃的氧化及进一步与CO2反应形成手性环碳酸酯的两步串联反应(图15)。框架中Keggin 型多金属氧酸盐[ZnW12O40]6-阴离子来源于不稳定的W10O324-与Zn2+在水热条件下的重组,高价的钨可实现对烯烃的氧化;Zn2+离子不仅作为框架节点也是Lewis 酸催化中心;氨基取代的联吡啶配体在支撑骨架的同时可增强对CO2的吸附捕集效果;咪唑基手性小分子催化剂通过与Zn2+离子配位排布于空腔,使手性产物EE 值得以保证。上述多种催化活性位点使该MOFs 材料能在温和条件下实现低碳烯烃经氧化成环并进一步与CO2形成手性环碳酸酯的两步串联反应。
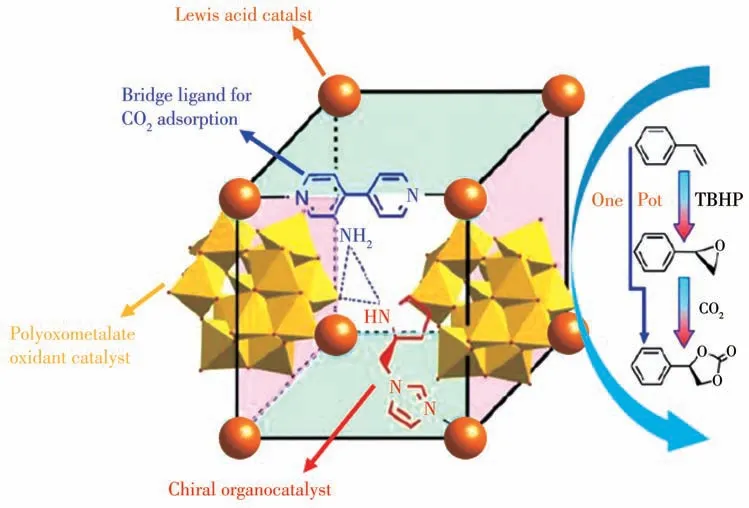
图15 多酸基MOF 13的构筑理念及其催化合成不对称环碳酸酯串联反应示意图[53]Fig.15 Design concept of achieving polyoxometalate based MOF 13 and tandem catalysis for asymmetric cyclic carbonate transformation[53]
2.2 功能化MOFs在CO2还原反应中的应用
近年来,CO2还原得到了广泛关注,还原的产物包括一氧化碳(CO)、甲烷(CH4)、甲醇(CH3OH)和甲酸(HCOOH)等[54-55]。均相催化剂通常催化活性高但需要在高压条件下进行(2~20 MPa),并且很难从催化系统中分离出来,催化剂不能循环利用[56-62]。相比较而言,MOFs 材料不但可以在单一材料中结合高反应性的Lewis 酸碱对来提高其催化能力,还可以利用其孔道结构限制和/或稳定具有催化活性的金属、金属氧化物、金属配合物、金属团簇等来增强其催化能力[63-64]。除此之外,单一MOFs 通过热解后可以在多孔碳基体中生成金属/金属氧化物纳米粒子,活性中心分散均匀,热稳定性和化学稳定性增强,从而实现在各种反应下高效转化CO2[65-66]。目前利用MOFs 催化剂实现CO2的还原反应包括光催化还原和电催化还原。
2.2.1 光催化CO2还原
利用光催化技术将太阳能转化为化学能,将CO2资源化利用的同时,也为减少温室气体排放提供了可能。MOFs 是一种功能化的有机-无机杂化材料,所以其具有类似于无机半导体的特性,同时与分子配合物一样具有较强的吸光能力和激发态寿命[67]。除此之外,MOFs 具有可捕获CO2分子、其有机配体容易修饰等优点,从而使其成为构建光催化CO2还原体系的理想材料(表2)。下面将根据MOFs 在光催化CO2还原体系的主要功能介绍近期的研究工作。
在光照下,MOFs 的有机配体受到光子的激发形成电子-空穴对,然后将激发态的电子传递给MOFs 材料中的金属节点(CO2还原的催化活性位点),最后金属节点将电子用于催化CO2还原生成各种还原产物,这些MOFs 就是光催化体系的光催化剂[68-70]。Li 等研究了以Ti 为金属节点的MIL-125(Ti)(14)以及氨基功能化的NH2-MIL-125(Ti)(15)的光催化CO2还原的活性。在以三乙醇胺(TEOA)为电子供体及质子源、乙腈为溶剂的体系中,MOF 15 比MOF 14 具有更高的催化CO2还原活性[71]。在10 h 的催化反应中,以MOF 15 为光催化剂的体系中生成还原产物甲酸8.14 μL,而MOF 14 为光催化剂的催化体系几乎没有甲酸生成(图16a)。在可见光激发下,MOF 15中有机配位体的电子从基态跃迀到激发态,然后转移给中心金属使Ti4+还原为Ti3+,最后Ti3+将电子传递到CO2分子生成还原产物甲酸,而Ti3+氧化为Ti4+,完成一次催化循环(图16b)。
基于铁的MOFs 材料具有强的吸光能力,因此经常被用作光催化剂[72-74]。2014 年,Li 等报道了一系列的基于铁MOFs 材料(MIL-101(Fe)、MIL-53(Fe)、MIL-88B(Fe))以及它们氨基功能化的MOFs,并将其作为光催化剂用于催化CO2还原[75]。在电子供体存在条件下,这些MOFs光催化剂还原CO2生成的产物都是甲酸。NH2-MIL-101(Fe)(16)光催化剂的催化活性最高,这是因为在该MOF 结构中有配位不饱和Fe。Fe3O 簇被光激发后,激发态电子传递给Fe3+形成Fe2+,然后还原CO2生成甲酸。这些MOFs 光催化剂氨基功能化后,催化CO2还原的活性有了显著的提高(图17a)。这是因为不但氨基功能化后的MOFs可以通过Fe3O 簇被可见光激发得到激发态电子,而且氨基功能化的有机配体也可以被可见光激发得到激发态电子,并用于催化反应(图17b)。催化反应后的MOFs 光催化剂晶体结构和化学性质都没有改变,说明这些MOFs光催化剂在催化CO2还原体系中具有良好的稳定性。
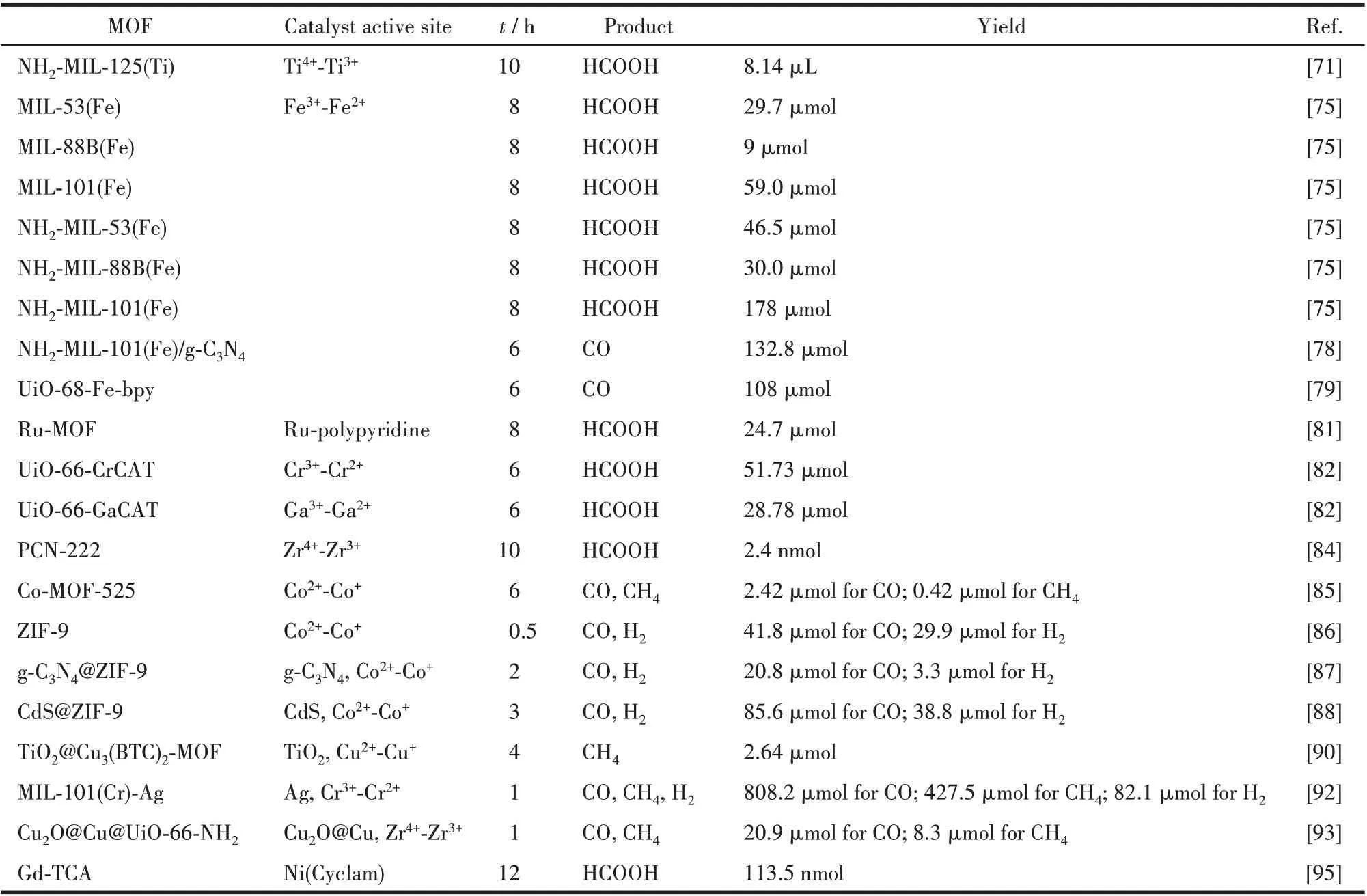
表2 一些功能化MOFs作为催化剂在光催化CO2还原反应中的性能Table 2 Catalytic performance of some reported functional MOFs catalysts in photocatalytic CO2 reduction
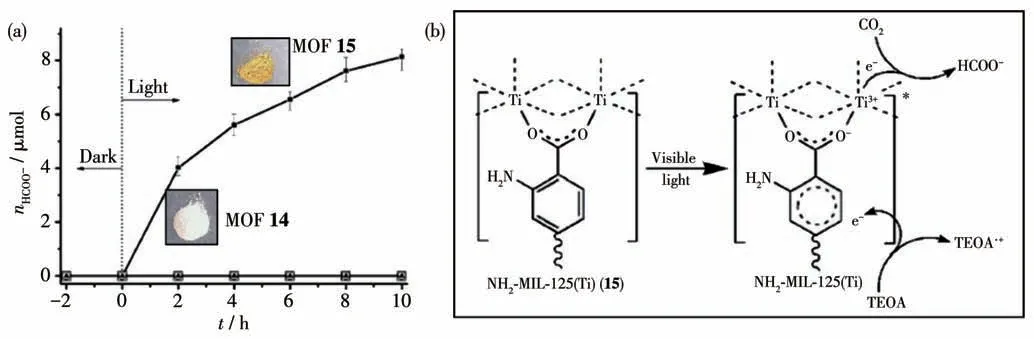
图16 (a)MOFs 14和15光催化CO2还原的活性;(b)MOF 15光催化CO2还原的可能机理[71]Fig.16 (a)Photocatalytic activity of CO2 reduction catalyzed by MOFs 14 and 15;(b)Proposed mechanism for photocatalytic CO2 reduction catalyzed by MOF 15[71]
近年来,我们课题组也探究了一系列铁基MOFs 作为催化剂光催化还原CO2。比如,我们利用铁基MOFs 作为光催化剂,实现了无溶剂体系中的光催化CO2还原,还原产物为CO[76];通过在NH2-MIL-101(Fe)中引入类石墨氮化碳(g-C3N4)形成NH2-MIL-101(Fe)/g-C3N4复合物实现光催化还原CO2为CO[77];首次实现在无溶剂条件下,以MIL-101(Fe)作为光催化剂光催化还原CO2为CH4[78]。紧接着,在2021 年我们课题组还利用bpy-CHO(bpy-CHO=2,2′-联吡啶醛)与UiO-68-NH2进行醛胺缩合反应合成了一种含有Fe(bpy)Cl3的CO2还原光催化剂UiO-68-Fe-bpy(17)[79],并证明该催化剂在可见光照射下,三乙醇胺(TEOA)为电子供体可以高效和高选择性地将CO2还原为CO(图18)。
除了MOFs 结构中的金属节点可以作为光催化CO2还原的催化活性位点外,对于以金属配合物为有机配体的MOFs 材料,其有机配体中的金属也可以作为光催化CO2还原的催化活性位点。如2011年,Lin 等[80]利用一系列Ir、Re 和Ru的配合物作为有机配体制备了相应的MOFs 材料M-UiO-67,其中M为Re (18)、Ir (19)和Ru (20)(图19)。在3 种MOFs 光催化剂中,MOF 18 具有最高的光催化CO2还原活性,且生成的还原产物CO 的量是Re 金属配合物的3倍。Luo等报道了{Cd2[Ru(dcbpy)3]·12H2O}n(dcbpy=4,4′-二羧酸-2,2′-联吡啶)的光催化剂Ru-MOF[81]。在以TEOA 为电子供体的光催化体系中,Ru-MOF将CO2还原生成甲酸,其光催化CO2还原的活性比大多数的MOFs 光催化剂高。这是因为Ru-MOF 具有很长的激发态寿命(5.49 μs),而有机配体[Ru(H2dcbpy)3]Cl2的激发态寿命要短很多(0.483 μs)。更长的激发态寿命意味着电子与空穴复合的速率更慢,这有利于电子传递给催化活性位点进行催化CO2还原反应。
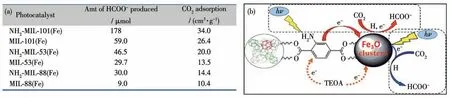
图17 (a)不同Fe基MOFs光催化CO2还原的活性;(b)MOF 16光催化CO2还原的可能机理[75]Fig.17 (a)Photocatalytic CO2 reduction activity of different Fe-MOFs;(b)Proposed mechanism for photocatalytic CO2 reduction catalyzed by MOF 16[75]
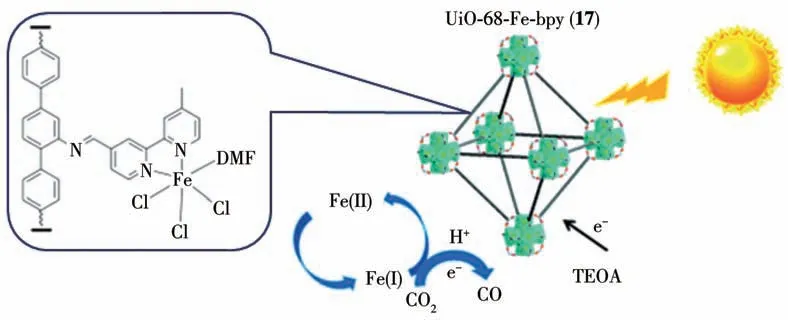
图18 MOF 17的结构及其光催化CO2还原反应示意图[79]Fig.18 Structure and schematic digram of MOF 17 for CO2 photoreduction reaction[79]
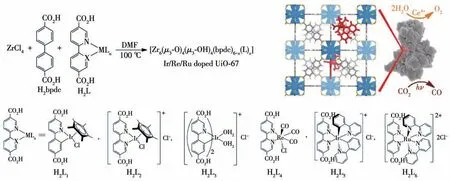
图19 M-UiO-67(M为Re(18)、Ir(19)和Ru(20))的合成及其配体结构[80]Fig.19 Structures of the ligands and synthesis of M-UiO-67(M=Re(18),Ir(19)and Ru(20))[80]
除此之外,Cochen 等[82]制备了羟基功能化的MOFs材料UiO-66-CAT,随后利用MOFs中的羟基基团分别与Cr 和Ga 进行配位得到了UiO-66-CrCAT(21)和UiO-66-GaCAT (22)两种MOFs 光催化剂(图20a)。在可见光条件下光催化反应6 h 后,MOF 21生成51.73 μmol 甲酸,MOF 22 生成28.78 μmol 甲酸,而UiO-66-CAT没有甲酸生成。这是因为在光催化反应过程中有机配位体充当光敏剂作用,在可见光激发下得到激发态电子,然后将激发态电子传递给Cr 或者Ga 催化CO2还原反应(图20b)。当光催化剂不含配位金属时,有机配位体不能将电子传递给节点中还原电位较负的金属Zr4+,所以UiO-66-CAT不能光催化CO2还原。
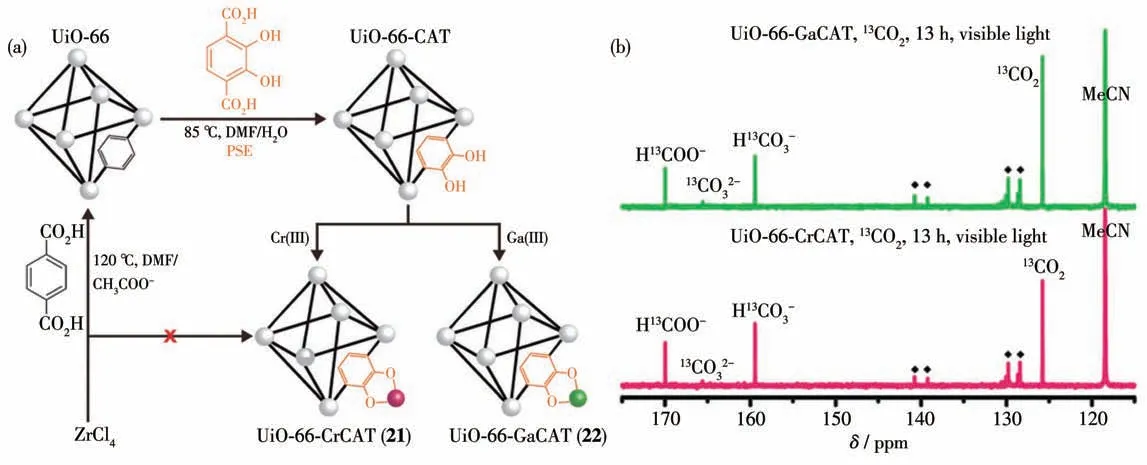
图20 (a)利用后修饰法制备MOFs 21和22光催化剂;(b)光催化剂MOFs 21和22催化CO2还原反应13 h后的13C NMR[82]Fig.20 (a)Preparation of MOFs 21 and 22 through post synthetic exchange(PSE);(b) 13C NMR spectra of photocatalytic CO2 reduction by MOFs 21(red)and 22(green)after 13 h[82]
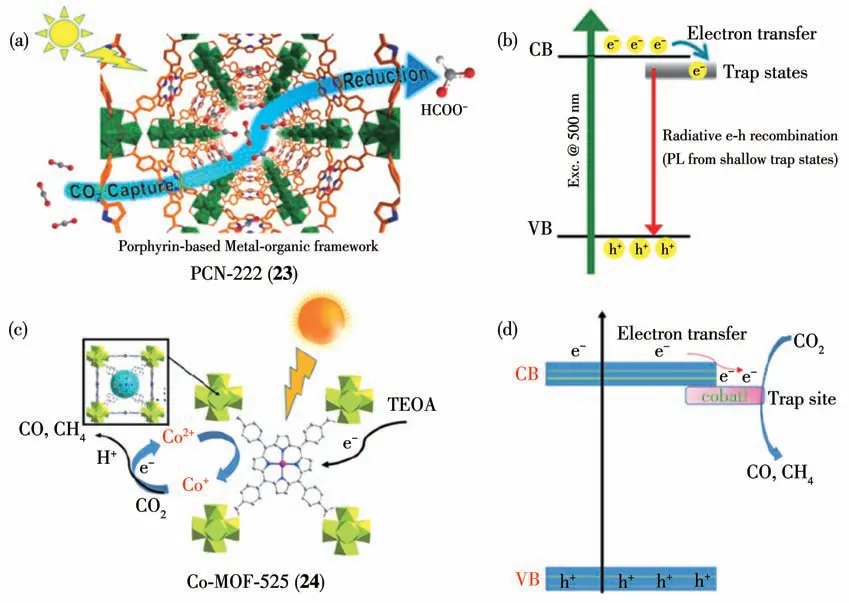
图21 (a)MOF 23的结构及其光催化还原CO2示意图;(b)MOF 23光催化还原CO2的催化机理;(c)MOF 24的结构及其光催化还原CO2示意图;(d)MOF 24光催化还原CO2的催化机理[84-85]Fig.21 (a)Structure of PCN-222(23)and schematic illustration of photocatalytic CO2 reduction process;(b)Proposed mechanism for photocatalytic CO2 reduction by MOF 23;(c)Structure of MOF 24 and schematic illustration of photocatalytic CO2 reduction process;(d)Proposed mechanism for photocatalytic CO2 reduction by MOF 24[84-85]
除了羧酸类金属配合物配体构筑的MOFs 可作为光催化剂实现CO2的光催化还原外,卟啉金属配合物也被用来光催化还原CO2[83]。如Jiang 等[84]选择了一种由卟啉四羧酸配体与锆离子构筑的MOF:PCN-222 (23)作为催化剂,在可见光照射下实现了光催化还原CO2为甲酸根离子(图21a)。在以MOF 23为光催化剂与TEOA作为电子供体的体系中光催化反应10 h,生成2.4 nmol 甲酸。光催化过程中MOF 23的有机配体卟啉被光激发,然后将激发态电子传递给Zr4+形成Zr3+,最后Zr3+将CO2分子还原成甲酸。作者还通过超快瞬态光谱和稳态/瞬态荧光光谱数据,证实MOF 23 骨架中存在一类长寿命电子陷阱态在有效抑制光生电子-空穴复合方面的微观动力学机制,从而揭示了该MOFs 材料光催化转化效率与光生电子-空穴分离效率之间的关系(图21b)。2016 年Ye 等[85]同样选择了一种卟啉四羧酸配体,在溶剂热条件下制备了由卟啉钴和Zr4+构筑的稳定Co-MOF-525(24)(图21c)。MOF 24 光催化还原CO2生成的主要产物为CH4和CO。由于MOF 24具有长的激发态寿命且对CO2吸附量大,所以该光催化剂表现出了很高的催化活性。MOF 24 中的有机配体Co-TCPP 的导带电位为-0.56 V(vs NHE),ZrO簇的导带电位为-1.09 V(vs NHE)。Co-TCPP 导带上的电子不能传递给ZrO 簇,所以Zr 不是催化CO2还原的催化活性位点,而Co才是催化活性位点。催化机理研究表明,MOF 24中的有机配Co-TCPP 受光激发将电子传递给催化活性位点Co2+形成Co+,然后在质子的参与下将CO2还原生成CH4和CO(图21d)。
某些MOFs 在光催化CO2还原体系中本身不能吸收光进行催化反应,需要光敏剂为其提供催化反应所需的电子,才能进行催化CO2还原反应,这些MOFs 就是光催化体系的催化剂。这类体系中光敏剂一般为光催化还原CO2的活性很低的半导体或者吸光染料,而MOFs 催化剂的加入可以显著地提高体系的催化活性。如Wang 等首次以沸石分子筛结构的MOF:ZIF-9 (25)作为催化剂、三联吡啶钌为光敏剂和TEOA 为电子供体构建了三组分光催化CO2还原体系[86]。在光催化反应0.5 h 过程中生成41.8 μmol CO 和29.9 μmol 氢气,CO 的选择性为58.3%。该体系的光催化活性比绝大数MOFs 光催化剂的体系活性高,但是该催化体系的稳定性差。光催化反应前30 min 生成CO 的速率最快,后面逐渐变慢,催化反应60 min 后CO 的量就不再增加,说明此时体系已经失活(图22a)。对催化反应后的MOF 25 进行表征发现其晶格结构以及化学性质都没有明显的变化,且MOF 25 在催化体系中进行多次循环光催化CO2还原反应,生成的还原产物没有明显的变化,这说明MOF 25 催化剂在光催化反应过程中没有失活。由于三联吡啶钌光敏剂在光催化反应过程中被破坏,从而使该体系失活。为了提高光催化体系的稳定性,该课题组用稳定的半导体材料g-C3N4取代三联吡啶钌作为体系的光敏剂,构建了g-C3N4与MOF 25 混合光催化体系[87]。在光催化反应2 h 后,生成20.8 μmol CO 和3.3 μmol 氢气,虽然该体系的催化活性有一定的下降,但是CO 的选择性(86.3%)却有了显著提高。光催化反应0~2 h CO 的生成速率基本没有变化,到3 h 才开始逐渐下降,说明用g-C3N4取代三联吡啶钌使得催化体系的稳定性有了明显提高(图22b)。随后该课题组用可见光吸收能力更强的半导体材料CdS 作为光敏剂,构建了CdS 与MOF 25 混合光催化体系[88]。催化反应1 h,生成50.4 μmol CO,催化反应3 h 后,生成85.6 μmol CO(图22c)。该体系在光催化CO2还原过程中的稳定性与g-C3N4+MOF 25 混合光催化体系相当,但是催化活性却有了明显提高。
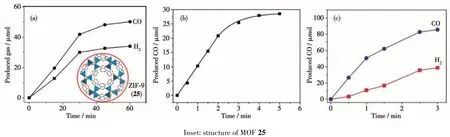
图22 MOF 25分别与三联吡啶钌(a)、g-C3N4(b)和CdS(c)组成的光催化CO2还原体系生成CO的量随时间变化的曲线[86-88]Fig.22 Time-production plot of CO produced in CO2 photoreduction system catalyzed by MOF 25 with Ru(bpy)32+(a),g-C3N4(b)and CdS(c),respectively[86-88]
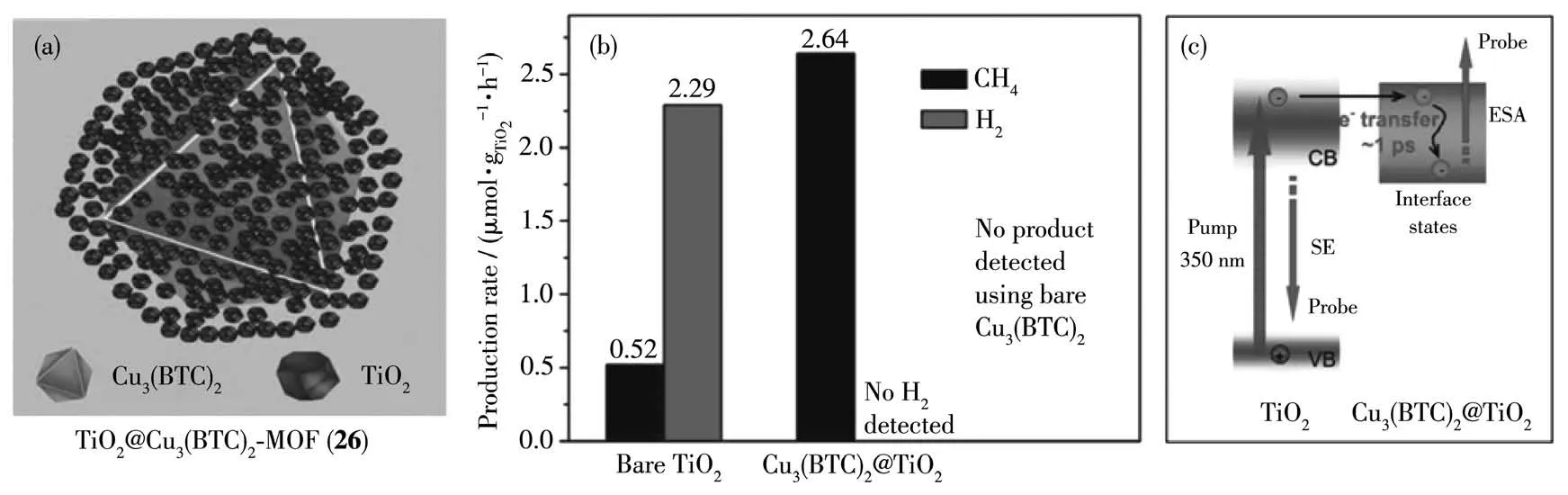
图23 (a)MOF 26复合催化剂的结构;(b)MOF 26复合催化剂的催化效率;(c)MOF 26复合催化剂的可能光催化机理[90]Fig.23 (a)Structure of MOF 26 composite catalyst;(b)Catalytic efficiency of TiO2 and MOF 26 composite catalyst;(c)Proposed mechanism for photocatalytic CO2 reduction by MOF 26 composite catalyst[90]
用半导体作为光敏剂与MOFs 催化剂组成的复合光催化CO2还原体系具有较高的稳定性[89]。比如Li 等[90]将TiO2负载到Cu3(BTC)2-MOF 表面上形成TiO2@Cu3(BTC)2-MOF (26)(H3BTC=1,3,5-苯三甲酸)复合催化剂(图23a)。TiO2在紫外光条件下可以将CO2还原为甲烷,形成MOF 26 复合催化剂后,光催化生成甲烷的速率有了显著的提髙,而单独的Cu3(BTC)2-MOF没有光催化CO2还原的活性(图23b)。其催化机理是TiO2首先将激发电子传递给Cu3(BTC)2-MOF,然后在Cu3(BTC)2-MOF 表面进行催化CO2还原反应。MOF 催化剂负载到半导体表面可以显著提高体系的光催化CO2还原活性(图23c)。但是目前对复合催化剂的催化过程还不是很清楚,所以深入研究复合催化剂催化CO2还原的机理是非常必要的。
近年来,我们课题组也利用功能化MOFs 和一系列纳米颗粒形成复合材料来提高CO2光催化体系的稳定性和活性[91]。比如,我们利用MIL-101(Cr)与Ag纳米颗粒结合得到MIL-101(Cr)-Ag复合材料实现光催化还原CO2,并通过调控MIL-101(Cr)的尺寸来进一步调控复合材料的催化活性[92];通过利用UiO-68 稳定Pt 纳米颗粒得到UiO-68-Pt 的复合材料实现光催化还原CO2,并通过调控复合材料结构中Pt 的含量和位置来调控复合材料的催化性能[93]。最近,我们利用简单的溶剂热合成了一例含有UiO-66-NH2和Cu2@Cu 的复合材料Cu2O@Cu@UiO-66-NH2(27),并证实该材料是立方结构[94]。三元纳米立方体结构使得该复合材料具有更高的可见光吸收能力和电荷载流子密度,同时促进光生电子的分离和迁移,最终提高了光催化CO2还原活性(图24)。
另外,如果MOFs 在催化体系中作为光敏剂而没有催化活性或者催化活性特别弱,当体系中加入催化剂后催化活性得到显著提髙。这一体系中MOFs 受光激发并将激发态电子传递给催化剂,CO2还原反应在催化剂表面进行。目前关于用MOFs 作为光催化CO2还原体系光敏剂的研究特别少。2016年,Wu 等[95]用以Gd 为金属节点的MOF 材料Gd-TCA(28)作为光敏剂,金属配合物Ni(Cyclam)作为催化剂与电子供体三乙胺(TEA)组成三组分光催化CO2还原体系(图25)。在光催化反应12 h后,生成甲酸113.5 nmol,同位素追踪实验证明甲酸中的碳来自CO2。Ni(Cyclam)催化剂在光催化CO2还原体系中选择性地生成CO,而该体系中生成的主要产物却是甲酸,所以该体系的催化反应机理还有待进一步研究[89-91]。

图24 MOF 27的结构和光催化CO2还原的示意图[94]Fig.24 Structure of MOF 27 and schematic illustration of photocatalytic CO2 reduction process[94]
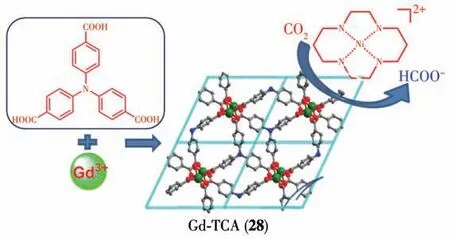
图25 MOF 28与Ni(Cyclam)光催化CO2还原的示意图[95]Fig.25 Representation of cooperation of MOF 28 and Ni(Cyclam)for photocatalytic reduction of CO2[95]
2.2.2 电催化CO2还原
电化学催化也是催化领域的一个重要分支。近年来,人们开始探索将MOFs 材料用于电化学过程,包括利用MOFs 材料电催化还原CO2[99-101]。一些功能化MOFs 在电催化CO2还原反应中已表现出很大的潜力(表3)。
2012 年,Kumar 等[102]使用MOFs 材料Cu3(BTC)2(29)作为催化剂用于电催化CO2还原(图26a)。MOF 29在有机相(DMF)体系中将CO2还原得到草酸,但是催化反应需要很高的过电势,且还原产物草酸的法拉第效率只有51%(图26b)。作者认为催化还原过程中CO2分子物理吸附在MOF 29 的孔隙中。MOF 29 中的Cuギ先被还原成Cuガ,然后Cuガ和CO2结合形成Cuガ-CO2中间体,MOFs 材料中的配体BTC起到路易斯酸的作用,可以稳定中间体。2 个Cuガ-CO2中间体聚合到一起,再进一步还原和质子化得到草酸。虽然MOF 29 催化活性较低且产物选择性也不高,但是该工作为开发更多用于电催化CO2还原的MOFs催化剂提供了基础。
2016 年,Kang 等[103]用溶剂热法制备得到了以H3BTC 为有机配体、Zn 为节点的金属有机框架材料Zn-MOF(30)。通过用电沉积的方法将Zn-MOF沉积到碳纸(CP)上得到Zn-MOF/CP 电极来进行CO2的电催化还原(图27a 和27b)。在离子液体体系中催化CO2还原得到的主要产物为甲烷,其法拉第效率大于80%,首次实现了在电催化体系中选择性生成CH4。用其他有机电解质取代离子液体或者用其他催化剂取代Zn-MOF/CP 电极,催化还原得到的主要产物都是CO,没有甲烷生成。这表明,只有在Zn-MOF/CP 电极和离子液体协同作用下才能生成甲烷。MOF 30 对CO2的吸附量最大,其次是CO,对甲烷的吸附量最小。表明吸附在MOF 30 表面的CO2先被还原成CO,由于MOF 30对CO具有较强的吸附作用,被吸附的CO 会进一步被还原成甲烷。作者推测了该体系的催化机理:离子液体的阳离子先吸附在MOF 30 表面,CO2被MOF 30 表面的离子液体捕获,CO2分子得到一个电子形成CO2·-中间体,然后再得到一个电子生成CO。在这步反应过程中,MOF 30 把电子传递给CO2分子,离子液体通过配位的方式使CO2·-中间体稳定。生成的CO 吸附在MOF 30 的表面,然后得到6 个电子和6 个质子被还原成甲烷(图27c)。
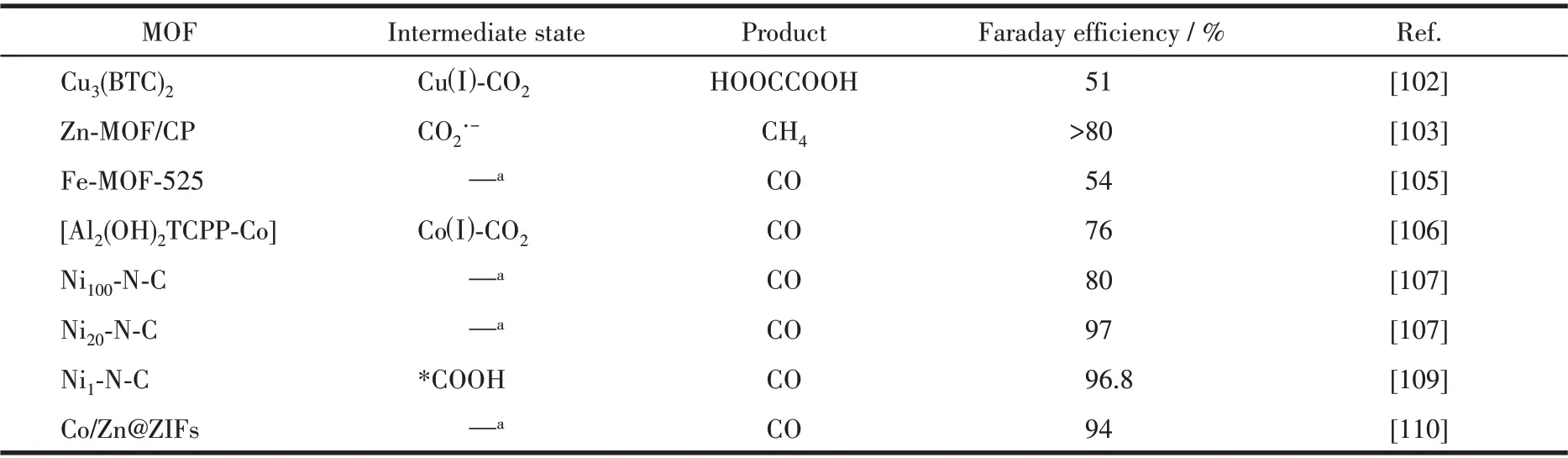
表3 一些功能化MOFs作为催化剂在电催化CO2还原反应中的性能比较Table 3 Catalytic performance of some reported functional MOFs catalysts in electroreduction of CO2
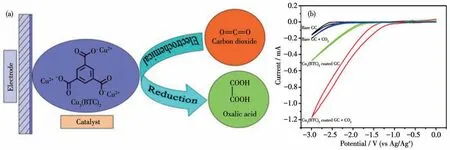
图26 (a)MOF 29电催化CO2还原示意图;(b)不同催化剂电催化CO2还原的循环伏安(CV)曲线[102]Fig.26 (a)Schematic diagram of electrocatalytic CO2 reduction by MOF 29 as catalyst;(b)Cyclic voltammetry(CV)curves of electrocatalytic CO2 reduction by different catalysts[102]
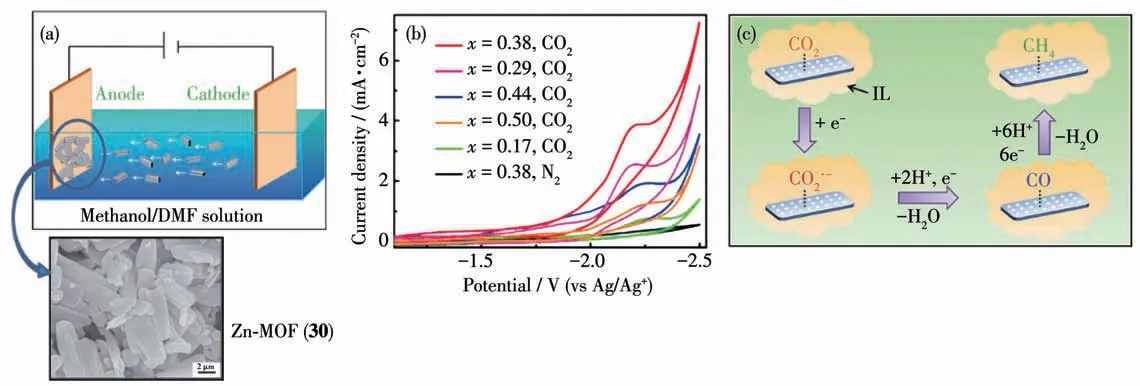
图27 (a)Zn-MOF/CP电极示意图及MOF 30的纳米结构;(b)Zn-MOF/CP电极电催化还原CO2的CV曲线;(c)Zn-MOF/CP电极电催化还原CO2生成甲烷的可能机理[103]Fig.27 (a)Schematic diagram of Zn-MOF/CP and nano-structure of MOF 30;(b)CV curves of electrocatalytic reduction of CO2 on Zn-MOF/CP electrode;(c)Possible mechanism of electrocatalytic reduction of CO2 to methane on Zn-MOF/CP electrode[103]
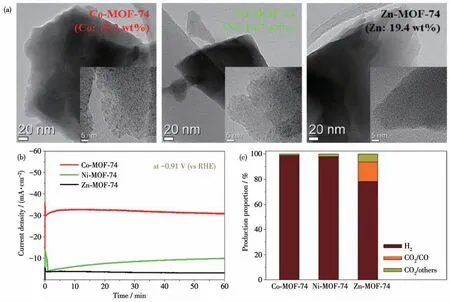
图28 (a)M-MOF-74(M=Co(31)、Ni(32)、Zn(33))的透射电镜(TEM)图;(b)在-0.91 V(vs RHE)电压下,60 min内催化剂M-MOF-74电催化CO2还原;(c)在-0.91 V(vs RHE)电压下,60 min后催化剂M-MOF-74电催化还原CO2的产物选择性[104]Fig.28 (a)Transmission electron microscope(TEM)images of M-MOF-74(M=Co(31),Ni(32),Zn(33))with magnified images(inset);(b)Electrocatalytic reduction of CO2 by M-MOF-74 at-0.91 V(vs RHE)for 60 min;(c)Selectivity of electrocatalytic CO2 reduction products by M-MOF-74 at-0.91 V(vs RHE)for 60 min[104]
2017 年,Choi 等[104]用2,5-二羟基对苯二甲酸作为有机配体与Co2+、Ni2+和Zn2+在溶剂热条件下自组装配位得到了一系列MOFs:M-MOF-74,其中M=Co(31)、Ni (32)、Zn (33)(图28a)。将M-MOF-74 分别负载到玻碳电极上作为工作电极,在0.5 mol·L-1KHCO3溶液及-0.91 V(vs RHE)电压下电催化CO2还原。催化反应进行60 min 后,3 种MOF-74 催化剂中,MOF 31 的电流密度远远大于MOF 32和MOF 33(电流密度最小)(图28b)。由于CO2还原产物的选择性和催化电流变化趋势刚好相反,MOF 31只生产了极其微量的CO,说明MOF 31 是一种比较好的析氢催化剂,而几乎没有催化CO2还原的活性。MOF 32也只得到少量CO,说明MOF 32 还原质子制氢活性较低,催化CO2还原活性更低。MOF 33 的CO2还原产物(CO 和液体产物)比例为21.3%,氢气为78.7%。这表明Zn-MOF-74(33)具有一定的催化CO2还原的活性。这是因为Zn 析氢的过电势较高,而还原CO2的过电势相对较低(图28c)。
由于MOFs 的三维网络结构并非共轭结构,不利于质子和电子的传导,限制了其在电催化还原CO2反应的催化活性。为了改善这一缺点,研究者在MOFs 的配体中连入具有大共轭结构的卟啉分子,来提高其质子或电子传导效率。如Hupp 等[105]就制备了基于卟啉铁(Fe-TCPP)的MOFs 薄膜Fe-MOF-525(34)(图29a)。MOF 34在以乙腈为溶剂的有机相体系及-1.3 V(vs NHE)电压下电催化反应5 h,其最大电流密度大约为2.3 mA·cm-2。当体系加入三氟乙醇(TFE)时,其最大电流密度增加到5.9 mA·cm-2,这说明TFE 可以为催化反应提供质子,从而提高体系的催化活性(图29b)。根据薄膜中活性催化剂的量计算得到CO 的TON 值为272,相对应的TOF值为64 h-1(图29c)。催化剂MOF 34在该体系中虽然有较高的电流密度,但是选择性却很低,CO 的法拉第效率只有54%,加入TFE后,虽然活性增加了,但是CO的法拉第效率却更低(41%)(图29d)。
同年,Yang 等[106]通过原子层沉积(ALD)技术得到了以卟啉钴Co-TCPP 为有机配体的MOF 薄膜,卟啉钴通过卟啉分子的4个羧酸基团和铝离子配位形成规则有序的MOF:[Al2(OH)2TCPP-Co] (35)。将MOF 薄膜作为催化剂在CO2饱和的KHCO3缓冲液(0.5 mol·L-1)中进行电化学催化CO2还原(图30a)。在-0.7 V(vs RHE)电压下电解7 h 后可以得到16 mL CO(0.71 mmol)(图30b)。ICP 分析表明MOF 35 电催化CO2还原的TON 为1 400,法拉第效率为76%,稳定后的电流密度大约为1 mA·cm-2。光谱电化学测试表明MOF 35 中的Coギ在-0.5 V(vs RHE)电位下被还原成中间态Coガ。该催化剂催化还原生成CO2的Tafel 斜率为165 mV·dec-1。根据以上结果,作者认为CO2分子和中间态Coガ形成Coガ-CO2是该体系催化反应的决速步骤,这与卟啉钴分子催化CO2还原的机理一致。
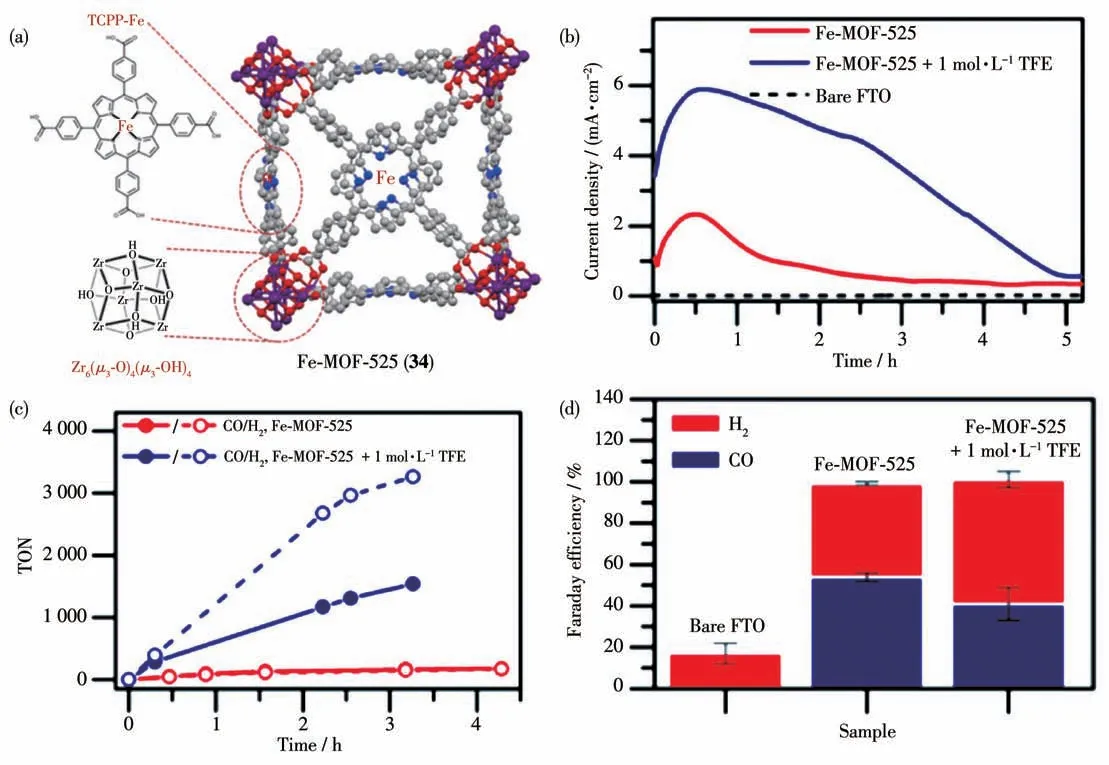
图29 (a)MOF 34的结构;(b)加入三氟乙醇前后MOF 34所需的电流密度随时间变化曲线;(c)加入三氟乙醇前后MOF 34的TON值随时间的变化曲线;(d)电催化反应的法拉第效率[105]Fig.29 (a)Structure of MOF 34;(b)Current density vs time for MOF 34 without(red)and with(blue)TFE,and a bare FTO blank(black);(c)TON vs time for MOF 34 without(red)and with(blue)TFE;(d)Faraday efficiency of the electrocatalytic reaction[105]
2020 年,我们课题组先是通过调控PCN-222 中卟啉配体TCPP 和卟啉镍配体Ni-TCPP 的比例合成了一系列具有不同Ni、N、C 比例的MOFs 前驱体,然后再热解腐蚀这些MOFs 前驱体得到含有不同比例金属镍的碳基镍催化剂Ni100-N-C (36)、Ni40-N-C(37)和Ni20-N-C(38)(图31a)。将36、37 和38 作为催化剂在CO2饱和的KHCO3缓冲液(0.5 mol·L-1)中进行电化学催化CO2还原[107]。催化剂36 在-0.7 V(vs RHE)电压下法拉第效率只有80%。在同样条件下,38 却显示出高达97%的法拉第效率,其催化还原生成CO 的Tafel 斜率为109 mV·dec-1(图31b 和31c)。而且通过高分辨透射电镜(HRTEM)对38 的表面进行分析发现,碳基镍催化剂38 表面没有相应的PCN-222 纳米颗粒,表明催化剂中镍纳米颗粒的低含量有助于提高催化剂的催化性能,这与之前文献报道的Ni-N-C 是主要的催化物种一致。碳基镍催化剂38 在-0.53 V(vs RHE)电压下的催化TOF 值可达159 s-1,并且可以在100 mA·cm-2电压下有效持续催化8 h。
随后,Jiang 等[108-109]采取同样的策略,利用卟啉配体TCPP 和含有不同金属离子的M-TCPP 得到了含有不同金属的碳基催化剂M1-N-C,其中M=Fe(39)、Co (40)、Ni (41)、Cu (42),并将其用于电化学催化CO2还原(图32a)。相比于碳基催化剂39、40 和42,41 的TOF 值可达11 315 h-1,其Tafel 斜率为98 mV·dec-1(图32b)。作者利用密度泛函理论(DFT)探究了不同单原子金属中心碳基催化剂电催化CO2还原的机理(图32c)。一般来说,电催化CO2还原生成CO 的步骤如下:第一步是CO2吸附;第二步是生成甲酸中间体(*COOH);第三步是生成*CO 和CO 解离出来(*代表吸附的中间产物)。其中第二步中*COOH 的形成被认为是所有M1-N-C 催化剂的催化速率决定步骤。
除了单原子催化剂,Wang等通过在不同温度热转化双金属Co/Zn 和ZIFs,成功制备了一系列氮掺杂的碳材料,而在这些碳材料中Co是以原子级分散的[110]。有趣的是,通过改变转化温度可以控制Co位点的N 的配位数。其中,含有Co-N2活性位点的催化剂更容易活化CO2并促进CO 中间体的生成。最终,在520 mV 的过电位下,催化剂表现出了超高的CO法拉第效率(94%)以及电流密度(18.1 mA·cm-2)。值得注意的是,具有最低N 配位数的Co 催化剂催化生成CO 的TOF 高达1.82×105h-1,超过了大多数报道的金属基催化剂。
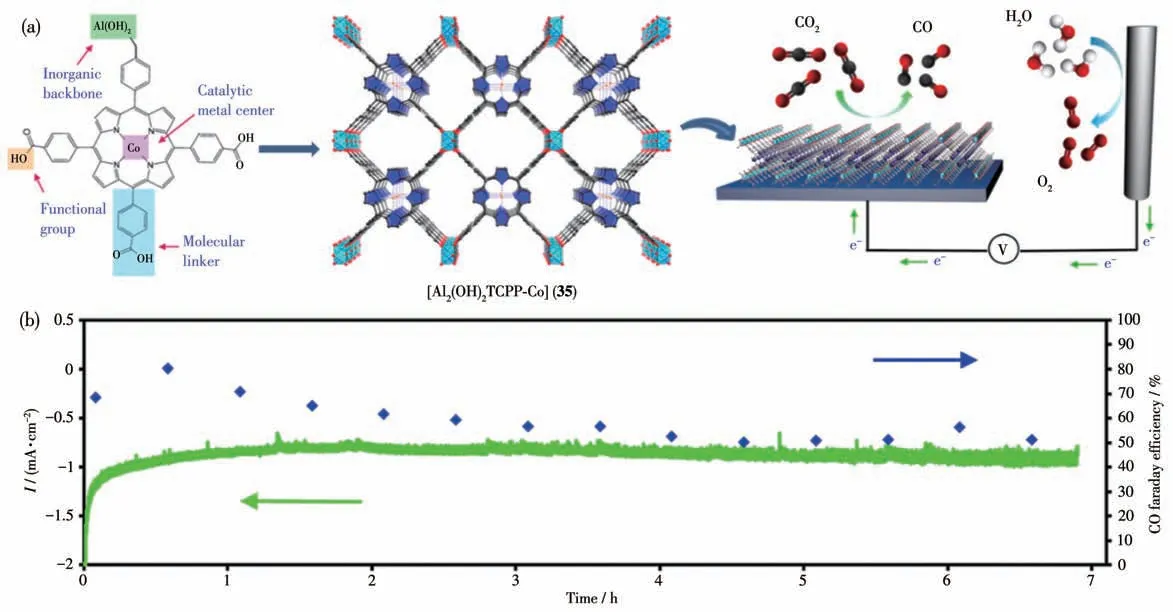
图30 (a)以MOF 35薄膜作为催化剂的电催化还原CO2示意图;(b)MOF 35电催化CO2还原的法拉第效率[106]Fig.30 (a)Schematic illustration of electrocatalytic reduction of CO2 using thin film MOF 35 as catalyst;(b)Faraday efficiency of MOF 35 as catalyst for electrocatalytic CO2 reduction[106]
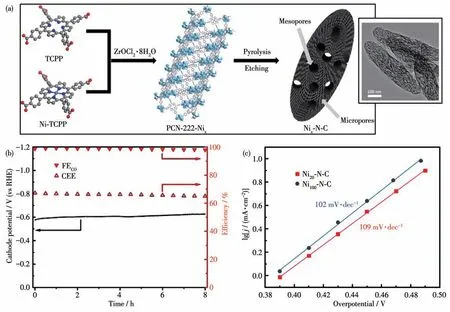
图31 (a)碳基镍催化剂Nix-N-C的制备过程及其TEM图;(b)催化剂38的法拉第效率随时间变化图;(c)催化剂36和38的Tafel斜率[107]Fig.31 (a)Illustration for fabrication of Nix-N-C catalysts and TEM image of the catalyst;(b)Time dependence of Faraday efficiency of catalyst 38;(c)Tafel slope for catalysts 36 and 38[107]
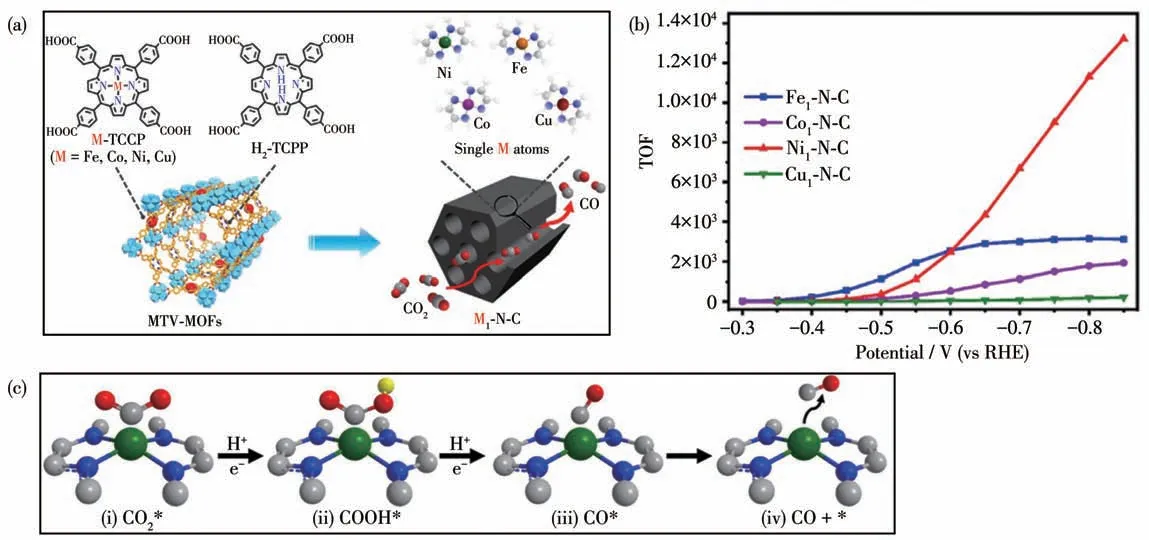
图32 (a)从M-TCPP到单原子催化剂M1-N-C(M=Fe、Co、Ni、Cu)的合成过程;(b)M1-N-C电催化还原CO2生成CO的TOF值;(c)利用DFT计算的M1-N-C电催化CO2还原可能的反应路径[109]Fig.32 (a)Illustration showing general fabrication of single-atom M1-N-C catalysts based on M-TCPP(M=Fe,Co,Ni,Cu)for electrocatalytic CO2 reduction;(b)TOF values of electrocatalytic reduction of CO2 to CO by M1-N-C;(c)Possible reaction path of electrocatalytic CO2 reduction by M1-N-C calculated by DFT[109]
3 总结与展望
MOFs 的功能化特性使其成为一类新型功能催化剂用于CO2的催化转化。本文从MOFs 的结构功能性入手,综述了近几年MOFs用于CO2催化转化的相关研究进展。无论是作为非均相催化剂来捕捉及化学固定CO2还是实现CO2光/电催化还原,MOFs都表现出优越的性能。这可以归结为以下几个因素:(1)相对较高的比表面积和多孔结构在稳定催化活性部分和防止失活的同时保证物质传导和电荷迁移;(2)各组分之间的协同效应可以活化CO2和底物分子促进CO2转化;(3) 基于MOFs 特定的组件,MOFs 复合材料和衍生物可以表现出更强的导电性、可见光吸收性等。虽然这些特性提高了MOFs在CO2的催化转化方面的潜在应用,但是目前该领域的研究还处于起步阶段,仍然有很大的空间来利用MOFs 实现CO2的催化转化。比如,CO2有机转化反应需要在高温高压条件下进行;CO2还原反应需要在碱性环境中进行;大多数MOFs 基催化体系仍然需要共催剂或电子牺牲剂等。
那么如何提高MOFs 催化剂的活性及稳定性,在更温和的条件下高效实现CO2的催化转化呢?首先,利用MOFs 确定的结构组分进行功能化[111-112]。比如:对合成MOFs 的有机配体修饰各种官能团来制备所需功能的MOFs催化剂;对MOFs结构中的金属或金属簇进行设计来提高材料的催化活性以及在MOFs 的空腔中引入具有催化活性的活性单元来实现材料的功能化等。其次,提高MOFs对CO2的吸附能力进而有效地提高催化体系对CO2的催化转化效率。比如:催化活性中心的局部CO2浓度的提高可以提高反应产率和选择性;高CO2吸附选择性MOFs 可以在混合气体或低CO2浓度或分压环境中具有高的催化活性。最后,运用各种先进表征手段对合成的MOF 基光催化剂进行详细表征,结合计算机模拟手段获取明确的MOFs 对CO2的催化转化反应机理,指导后续功能催化剂的设计合成等。
总之,功能化MOFs对CO2的催化转化已经显示出良好的应用前景。相关研究不仅对拓展MOFs 的功能应用具有重要意义,更是对绿色化学做出了贡献。相信上述难点和挑战会激发更多的科学家开展更多更深入的研究来促使这一领域更快、更好的发展。
猜你喜欢
中国有色金属学报(2018年2期)2018-03-26 07:58:37
中南大学学报(自然科学版)(2016年2期)2017-01-19 07:37:25
通信电源技术(2016年6期)2016-04-20 06:21:22
中国资源综合利用(2016年7期)2016-02-03 03:00:13
淮南师范学院学报(2015年3期)2015-03-22 01:16:19
河北科技大学学报(2015年5期)2015-03-11 16:16:34
无机化学学报(2014年12期)2014-02-28 17:33:51
无机化学学报(2014年4期)2014-02-28 17:31:23
无机化学学报(2014年3期)2014-02-28 17:30:48
应用技术学报(2014年1期)2014-02-28 14:52:11
