
铁掺杂对于二氧化锰催化剂表面吸附机理的理论研究*
2021-07-02 00:36:56任富忠潘开进王天浩
材料研究与应用 2021年2期
任富忠,冯 伟,潘开进,王天浩
铜仁学院 材料与化学工程学院,贵州 铜仁 554300
燃煤烟气排放时携带的重金属汞是大气汞污染的主要来源之一,汞在生态系统中的循环会对人类健康造成严重的伤害[1-2].煤燃烧过程中释放汞的主要途径有以下三个[3]:颗粒态汞、氧化态汞和单质汞.由于Hg0具有挥发性强、微溶于水且化学性质相对稳定的特性,难以采用常规的液相吸收方法去除.目前常用的Hg0的处理方法主要是:先采用选择性催化氧化脱汞法(SCR)工艺,将其先氧化为易溶于水的Hg2+,然后再液相吸收去除.催化氧化反应进行时主要涉及反应物间的电荷转移,这就需要脱汞催化剂的活性成分必须是具有活性较高的d电子或s电子才行,金属锰和金属铁外层分别具有7个活性电子和8个活性电子,使它们在催化氧化反应过程中可通过不同价态之间的转换实现电荷的转移,使其可与烟气中的氧配合完成Hg0向Hg2+的转换.在锰的氧化物中,MnO2因具有较高的汞脱除效率、较好的再生能力及不易失活等优点而备受关注[4-6].Fe2O3也由于具有易得、易分离、成本低及无二次污染等优点,在烟气脱汞领域中备受重视[7-9].煤炭中赋存的氯元素有助于Hg0的吸附氧化脱除[10-11],Hg0吸附在氯化活性位上并被不完全氧化成HgCl,气相HgCl可以进一步被氯化活性位完全氧化成HgCl2.
尽管已有众多的锰基催化剂被验证有良好的脱汞效果,但是对其脱汞机理的研究尚不全面,而采用计算机模拟的方法可以为新型催化剂的设计及催化机理的研究提供参考.采用基于密度泛函理论的第一性原理方法,研究了金属Fe掺杂MnO2(110)表面对于Cl,Hg和HgCl三种吸附物质的吸附状态,分析和对比三种吸附物质被吸附到铁掺杂的二氧化锰表面后的吸附能、键长和电荷分布状态,其模拟计算结果可为MnO2掺杂体系特性的实验研究提供理论指导.
1 第一性能原理及Materials Studio软件简介
第一性能原理是一个计算物理或计算化学专业名词,广义的第一性原理计算指的是一切基于量子力学原理的计算.量子力学计算就是根据原子核和电子的相互作用原理,计算分子结构和分子能量(或离子),从而计算物质的各种性质.广义的第一原理,包括以Hartree-Fock自洽场计算为基础的abinitio从头算和密度泛函理论(DFT)计算两大类.利用计算机模拟的方法,对实验中发现的某种新现象在理论上得到合理的解释,通过理论结果可以修正原有实验方法或进一步实现理论上的预测.所以,以第一性原理为代表的计算机模拟是当今实验研究必不可少的重要手段[12-14].
Materials studio(MS)软件是一款由美国Accelrys公司开发的材料科学软件,其综合应用了计算化学的量子力学法和分子学法,以及分子模拟的蒙特卡洛和分子动力学模拟方法.Materials Studio包含很多模块,是一个包括量子力学、介观模型、分子力学、分析工具模拟和统计相关在内的可视化建模环境.CASTEP 是美国剑桥大学凝聚态理论研发组开发的一款基于密度泛函理论的先进量子力学程序,是Material Studio软件的重要模块之一,通过整合使用这些先进理论和计算方法,MS 软件实现了对材料进行不同粒子尺寸和时间范围内的评估和计算,通过对各种小分子、纳米团簇、晶体、非晶体,以及高分子材料的性质进行深入的挖掘,为实际的科学研究提供了切实可靠的实验数据.
2 计算模型及方法
选取2×2×1的MnO2超晶胞进行计算,MnO2(110)的结构模型由连续的O—Mn—O结构组成,Fe掺杂前后的超晶胞如图1所示.从图1可见,结构中用一个Fe原子替换体系中间位置的目标Mn原子,以减少边界效应带来的影响,为了得到更加稳定而真实的结构,对各模型进行了几何优化至收敛,目的是使模型结构尽可能地与真实结构相接近,因此研究Fe掺杂MnO2后的吸附能、电荷及键长键角对于精确分析是非常有必要的.本文设定自洽场能量收敛标准为1.9×10-3kJ/mol,优化收敛标准的能量小于1.0×10-3eV,最大力设置为0.5 eV/nm,公差偏移2.0×10-4m,应力偏差0.1 GPa.
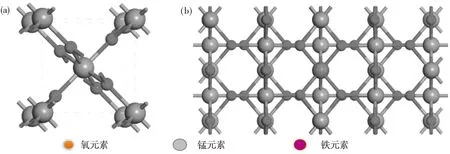
图1 纯MnO2和Fe掺杂MnO2结构模型Fig.1 Structural model of pure MnO2 and Fe doped MnO2(a)MnO2 原胞结构;(b)Fe掺杂的MnO2结构(a)MnO2 cell structure;(b)Fe doped MnO2 structure
3 吸附能的计算
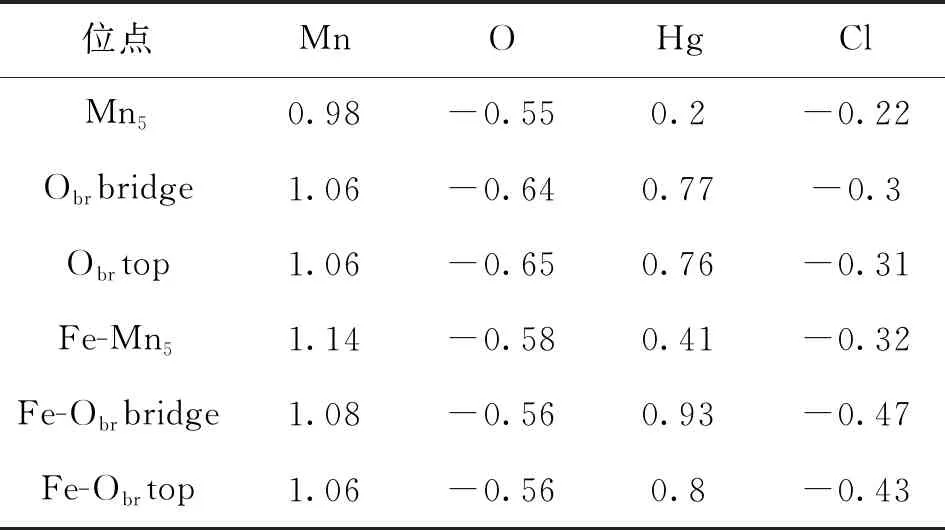
MnO2晶体最稳定的晶面是(110)面,MnO2(110)表面上有4中不同的原子,分别为Obr,Os,Mn5和Mn6,其中Obr连接两个Mn6,而Os分别连接Mn5和Mn6原子,考虑了MnO2(110)表面六种不同的高对称性吸附位,分别为Ostop,Obrtop,Mn5top,Obrbridge,Mn5bridge和hollow位,如图2所示.用3个O—Mn—O原子结构(9个原子层)来模拟MnO2(110)表面.通常,周期性平板模型的可靠性对研究结果至关重要,选择了底部5层固定在它们的主体位置,而允许顶部原子自由移动.采用2×2×1原胞优化Fe的结构,吸附能E(ads)采用如下方法计算:E(ads)=E(adsorbate-substrate)-E(adsorbate)-E(substrate),其中E(adsorbate-substrate)代表吸附后整个体系的总能量,E(adsorbate)代表吸附质的能量,E(substrate)代表吸附基底MnO2(110)的能量.E(ads)为负值表明吸附过程放出能量,其值越小表明吸附越强.吸附能计算值为负时,表明发生放热反应且吸附后体系更加稳定.E(ads)越小表示体系的稳定性越强,反之E(ads)越大表示体系的稳定性越差.吸附能满足 0.62 图2 MnO2(110)面上6个不同的吸附位置Fig.2 Six different adsorption sites on the MnO2 plane 表1和表2分别为MnO2(110)表面在Fe掺杂前后Cl吸附在不同位点是各种键长、吸附能及表面电荷的数据.由表1可以看出:从Fe掺杂前后的吸附能在-2.221~-1.546 eV之间,说明Cl在 MnO2(110)表面吸附属于化学吸附;对比Fe掺杂前后Mn—O键长变化发现,Fe掺杂后键长变大,表明Fe掺杂后二氧化锰晶胞体积增大,晶胞比表面积 表1 Fe 掺杂前后Cl吸附到MnO2表面不同位点的键长和吸附能Table 1 The bond length and adsorption energy of Cl adsorbed to different sites on the surface of MnO2before and after Fe-doping 增加可以提供更多的活性位点并增加电子迁移率,有助于提升二氧化锰的活性;从Cl—O键的吸附能和和键长数据可以看出,Fe掺杂前后Cl在MnO2(110)表面的最优吸附点都是Obrtop;从Cl—Mn键的吸附能和和键长数据可以看出,Fe掺杂后吸附能更低,键长更大,活性增强. 从表2可以看出,Fe掺杂后当Cl吸附在Obrtop 位点时,对Mn和O附近的电荷密度有明显影响,Mn附近的电荷密度减小,O附近的电荷密度增加,Cl附近的电荷密度也增加,说明Fe掺杂后O原子 表2 Fe 掺杂前后Cl 吸附到MnO2(110) 表面不同位点的电荷量Table 2 The charge amount of Cl adsorbed to different sites on the surface of MnO2(110) before and after Fe-doping 周围的电负性增加,Cl的电负性也增加,也说明Fe掺杂后Cl也是优先吸附于O位点上,二者形成的键较为稳定. 表3和表4分别为MnO2(110)表面在Fe掺杂前后Hg吸附在不同位点是各种键长、吸附能及表面电荷的数据.由表3可以看出:Fe掺杂前后Hg吸附到MnO2(110)表面都属于物理吸附,Fe掺杂后Hg在Mn吸附位点和O吸附位点的吸附能都增大,且Fe掺杂后Hg在O吸附位点的吸附能小于Hg在Mn吸附位点的吸附能,表明掺杂后Hg优先吸附在O上;掺杂后当Hg吸附在Mn吸附位点上时Mn—Hg键和Hg—O键键长都增大,而当Hg吸附在O吸附位点上时Mn—Hg键和Hg—O键键长都减小,表明Hg优先吸附于O吸附位点上. 表3 Fe掺杂前后 Hg吸附到MnO2(110) 表面不同位点的吸附能和键长Table 3 Adsorption energy and bond length of Hg adsorbed to different sites on the surface of MnO2(110) before and after Fe-doping 表4 Fe掺杂前后 Hg吸附到MnO2(110) 表面不同位点时原子的电荷量Table 4 Atomic charge amount of Hg adsorbed to different sites on the surface of MnO2(110) before and after Fe-doping 由表4可以看出,Fe掺杂后二氧化锰向Hg,Mn及O原子的电荷均有变化,原子彼此间的交互作用会对二氧化锰与单Hg原子间的电荷转移产生影响.Fe掺杂后当Hg吸附于Mn位点时,Hg原子电荷量减小,而Mn与O原子的电荷量增大,表明此时Hg与Mn及O原子之间存在协同作用;当Fe掺杂后当Hg吸附于O位点时,主要存在Hg与O之间的作用. 表5和表6分别为MnO2(110)表面在Fe掺杂前后HgCl吸附在不同位点是各种键长、吸附能及表面电荷的数据.由表5可以看出:Fe掺杂前后HgCl吸附在Mn位点时属于物理吸附,而吸附在O位点时属于化学吸附,并且优先吸附在Obrbridge位点上;掺杂前后当HgCl吸附在Obrbridge位点上时由于Cl—O键的键长最短,结合力最强,并且掺杂后Hg—O键的键长比掺杂前更短,表明Fe掺杂有助于促进HgCl与O吸附位点的相互作用. 表5 HgCl吸附到MnO2(110)表面在Fe掺杂前后的吸附能和键长Table 5 Adsorption energy and bond length of HgCl adsorbed to MnO2(110) surface before and after Fe-doping 由表6可以看出,Fe掺杂后Mn,O,Hg和Cl原子的电荷都有变化.当HgCl吸附在O位点上时,O原子的电荷减少,而Cl原子由于电负性和Hg原子的电负性较大,都从O原子上得到电子,且在吸附在Obrbridge位点上减小最明显,也证明HgCl优先吸附在Obrbridge位点上;HgCl和二氧化锰之间的电荷转移越多,二氧化锰对HgCl的吸附越强. 表6 HgCl吸附到MnO2(110)表面在Fe掺杂前后的电荷Table 6 The charge of HgCl adsorbed to the surface of MnO2(110) before and after Fe-doping 通过对Cl,Hg和HgCl在Fe掺杂前后MnO2(110)面的吸附作用,进行基于第一性原理的计算机模拟发现,Fe掺杂前后Hg吸附到MnO2(110)表面都属于物理吸附作用,而C和HgCl都属于化学吸附作用,而且三种吸附质都是优先吸附在MnO2催化剂的O吸附位点上.Fe掺杂MnO2可调节体系吸附能,从而改善MnO2的催化氧化性能.金属掺杂对于改善MnO2的催化氧化效果,具有参考意义.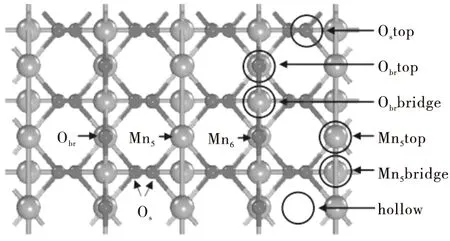
4 Fe掺杂MnO2(110)表面的吸附
4.1 Cl在Fe掺杂MnO2(110)表面的吸附
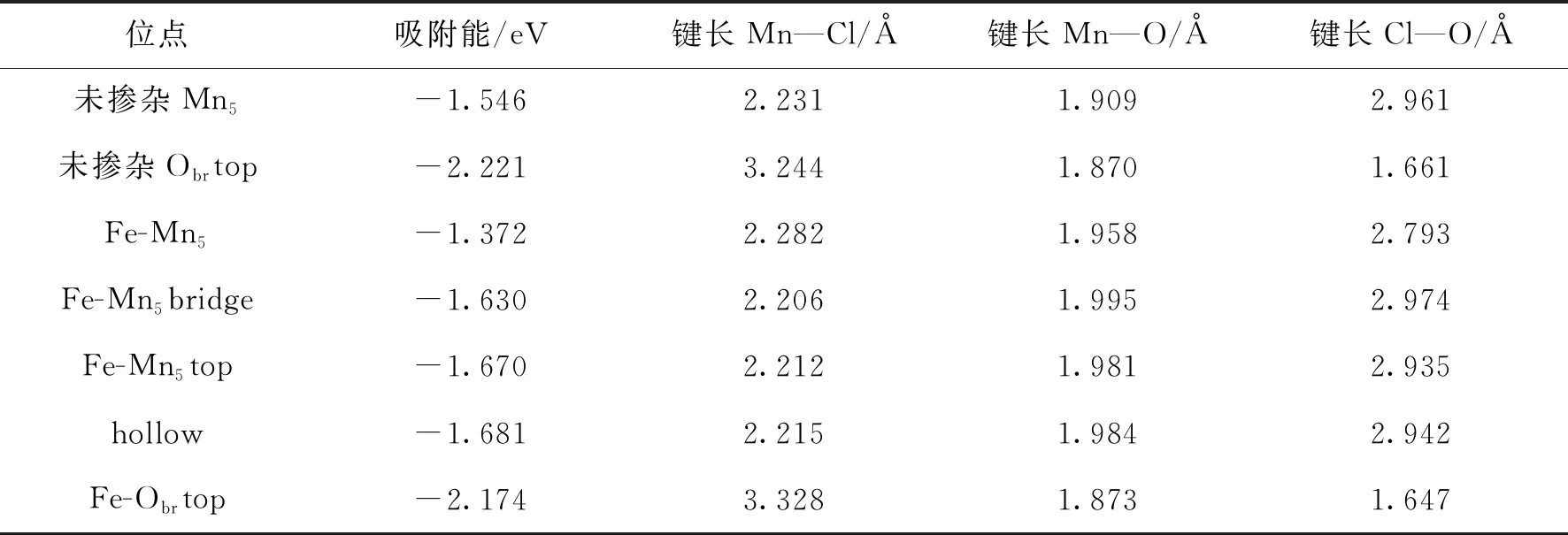

4.2 Hg在Fe掺杂MnO2(110)表面的吸附
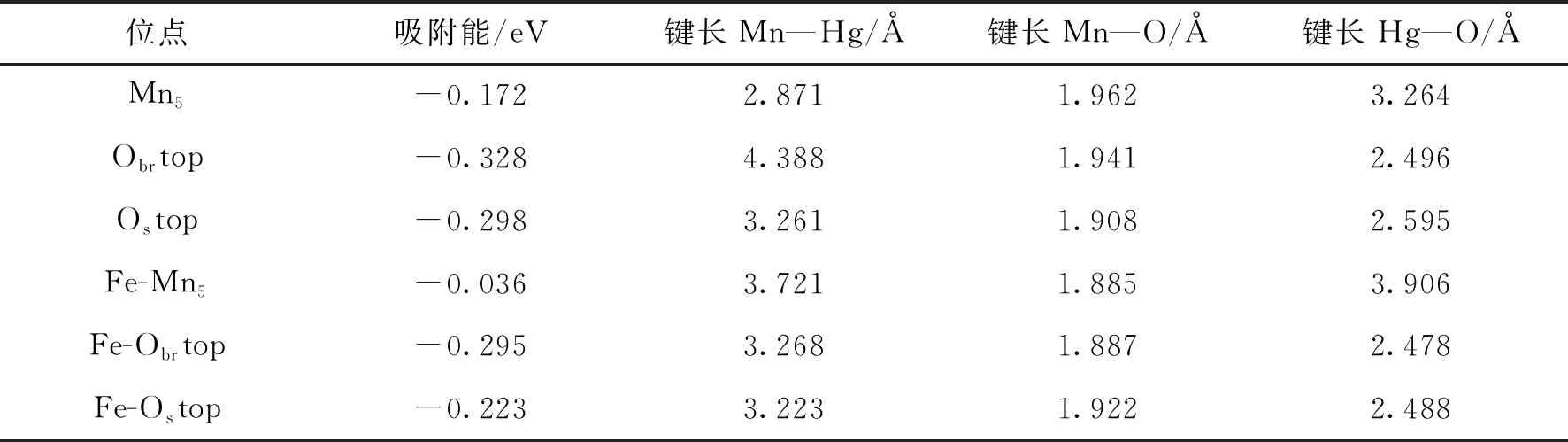

4.3 HgCl在Fe掺杂MnO2(110)表面的吸附

5 结 语
猜你喜欢
物理通报(2024年4期)2024-04-09 12:41:28
农村青少年科学探究(2022年3期)2022-05-13 07:50:56
中学生数理化·中考版(2021年10期)2021-11-22 07:26:40
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27 03:07:02
中国金属通报(2020年20期)2020-03-27 07:19:52
青岛大学学报(工程技术版)(2019年2期)2019-09-10 07:22:44
池州学院学报(2017年3期)2017-10-16 01:38:36
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
中学化学(2015年8期)2015-12-29 07:32:44
