
A位缺陷对(Pr0.5Sr0.5)1-xFe0.9Ru0.1O3-δ(0≤x≤0.2)阳极材料电化学性能及稳定性影响的研究*
2021-07-02 00:36覃铭霞杨洪宇王子鸣刘太楷杨成浩
材料研究与应用 2021年2期
覃铭霞,杨洪宇,谭 婷,王子鸣,宋 琛,刘太楷,刘 敏,杨成浩
1.华南理工大学 环境与能源学院,广州 510006;2.广东省科学院新材料研究所,现代材料表面工程技术国家工程实验室,广东省现代表面工程技术重点实验室,广东 广州 510650
利用活性金属纳米粒子对氧化物载体进行修饰是一种提高材料性能的常用方法,可广泛应用于光催化、热催化和电化学催化等领域中.其中负载金属纳米粒子的尺寸、粒径分布、元素组成及其与氧化物载体的相互作用,是决定催化活性、催化选择性及稳定性的关键.在固体氧化燃料电池(SOFC)中,这种改性方法已成功应用在多种类型的氧化物阳极上[1-4].近年来,随着对钙钛矿氧化物研究的深入,发现在还原气氛条件下部分钙钛矿材料的B位还原性金属可以被还原并析出至材料表面而形成活性纳米颗粒(NPs),对材料的导电性能与催化性能有显著影响,这为钙钛矿阳极的改性提供了一种有效的方法[5-6].催化活性金属元素在最初合成过程中被掺杂到氧化物宿主的晶格中,然后经还原处理后部分或全部金属物种将从晶格中析出,在氧化物载体上形成均匀分布的金属纳米粒子[7],其析出过程可由以下反应式表示[8]:
对于化学计量比A/B=1的ABO3型钙钛矿氧化物,即使在典型的高反应温度下也只能析出有限的B位过渡金属,如(La,Sr)MnO3,(La,Sr)CrO3和(La,Sr)TiO3[9].从化学反应式中可以看出,B位过渡金属元素的析出通常伴随着A位阳离子从化学计量的ABO3中分离,趋于形成AO相,但为了使材料能够保持热力学稳定的ABO3钙钛矿结构,反应的正向进行会受到限制.引入A缺陷是减少AO相形成和促进B位金属析出的一种有效方法.首先,A位缺陷(A/B<1)会使B位金属离子Mn+到M0的吉布斯自由能变得更负,从而促进成核过程.此外,随着A位缺陷的出现,还原环境中引入的氧空位很容易破坏钙钛矿晶格的稳定,在热力学驱动下B位金属离子自发地发生表面析出,以恢复化学计量比A/B=1[10].
此外,部分立方钙钛矿氧化物会在还原后转变为Ruddlesden-Popper层状钙钛矿氧化物.当占据B位的主要元素为还原稳定性较低的Fe,Cu和Co时,在还原气氛中处理后材料表面会生成大量的金属NPs,会出现立方相(ABO3)与层状相(Ruddlesden-Popper,RP)共存或完全转变成层状相,如K2NiF4型的Pr0.8Sr1.2(Co,Fe)0.8Nb0.2-O4+δ[5].层状相可以看成是由ABO3立方钙钛矿和AO岩盐层交替排列组成的,其化学式可以表示为An+1BnO3n+1[11-12].在B位金属与氧形成的八面体单元中,可变价金属的氧化还原可为电荷传输提供通道,岩盐层中的氧离子对可能产生的碳沉积直接氧化,使材料在碳氢燃料中具有较好的稳定性.目前,针对A缺位的研究一般应用于还原后不发生相变的钙钛矿氧化物中[13-14].当A缺位应用于还原后发生相转变的钙钛矿氧化物时,材料在还原过程中的结构变化及电化学性能并未有相关报道.
以Pr0.5Sr0.5Fe0.9Ru0.1O3-δ钙钛矿氧化物为研究对象,通过在材料合成过程中引入A缺位,研究了其在还原气氛中的析出过程及材料结构演变机理.同时,以加湿H2及C3H8-N2为燃料,探索了其作为SOFC阳极材料的电化学性能及运行稳定性.
1 实验部分
1.1 电极粉末制备
通过甘氨酸-燃烧法合成Pr0.5Sr0.5Fe0.9Ru0.1O3-δ(PSFR0),(Pr0.5Sr0.5)0.9Fe0.9Ru0.1O3-δ(PSFR0.1)和(Pr0.5Sr0.5)0.8Fe0.9Ru0.1O3-δ(PSFR0.2)材料.首先按化学计量比称取相应的Pr(NO3)3·H2O,Sr(NO3)2和Fe(NO3)3·9H2O加入烧杯中,加入去离子水并通过搅拌使其溶解,然后加入相应符合化学计量比的Ru(NO)(NO3)x(OH)y(x+y=3)、甘氨酸(与金属离子摩尔比为2∶1).将烧杯置于磁力搅拌器上,在100 ℃下加热搅拌1 h,再调高温度到200 ℃继续加热,使烧杯的水分蒸发至原料呈粘稠状,随后将温度设置为500 ℃进行加热直至原料完全自燃烧,待烧杯自然冷却至室温后将黑色前驱体粉末收集至刚玉坩埚,并将其置于800 ℃和空气气氛下煅烧3 h,从而获得钙钛矿结构的PSFR0,PSFR0.1和PSFR0.2.最后,将煅烧后的粉体用研钵研磨成较细的电极材料成品.阴极材料为La0.6Sr0.4Co0.2Fe0.8O3-δ(LSCF),通过甘氨酸-燃烧法合成.
1.2 电池制备及电化学性能测试
按照质量比1∶9称取聚乙烯缩丁醛(PVB)和松油醇,将PVB加入到松油醇中,然后将原料置于60 ℃烘箱中,待PVB完全溶解后得到质量分数为10%的PVB-松油醇粘结剂.制备阳极浆料时,先分别将PSFR0,PSFR0.1,PSFR0.2与Ce0.9Gd0.1O2-δ(GDC)按照质量比5∶5的比例加入到玛瑙研钵中[15],然后再加入粉体总质量5%的玉米淀粉,研磨均匀后加入与粉体质量比为1∶1的PVB-松油醇,反复研磨得到均匀的PSFR0-GDC,PSFR0.1-GDC和PSFR0.2-GDC阳极浆料.同样,制备阴极浆料时将LSCF与GDC按照质量比6∶4的比例加入到玛瑙研钵中,然后再加入粉体总质量5%的玉米淀粉,研磨均匀后加入与粉体质量比为1∶1的PVB-松油醇,反复研磨得到均匀的LSCF-GDC阴极浆料.
测试用的扣式电池均为La0.8Sr0.2Ga0.83Mg0.17O3-δ(LSGM)电解质支撑的单电池,LSGM电解质片通过干压法制备.在制备全电池和对称电池过程中,将浆料均匀地刷涂在电解质片上,其中阳极浆料完全覆盖电解质片,阴极浆料只刷涂在中心圆形部分,圆形直径为5 mm,得到的扣式电池的有效面积约为0.2 cm2.在全电池制备时,阳极为PSFRx-GDC,阴极为LSCF-GDC.由于阳极和阴极的煅烧温度不同,因此需要两步烧结.先在阳极刷涂PSFRx-GDC浆料,在1100 ℃下烧结2 h,接着在阴极刷涂LSCF-GDC浆料,然后将电池置于马弗炉中在1000 ℃下烧结2 h.在对称电池制备时,两侧电极均为PSFRx-GDC,两侧刷涂完毕后置于马弗炉中,在1000 ℃下进行一次烧结2 h.随后,在烧结完成的电极表面涂少量Pt浆作为电荷收集器.
采用四电极法进行电化学测试,以消除导线带来产生的欧姆电阻干扰.采用Zennium电化学工作站测试电池性能,阻抗扫描频率范围为100 KHz~0.1 Hz,电压扫描范围为1.1~0.2 V.燃料气由一根细陶瓷导管输送到阳极侧,反应产物及剩余燃料气经大陶瓷管的另一端排出.测试过程中,根据测试需要通入50 ml/min的加湿氢气(含3%的H2O)和40 ml/min的加湿C3H8(使用N2为载气,C3H8与N2体积流量比为1∶1)作为燃料气,阴极以空气中的氧气为氧化剂.
2 结果与讨论
2.1 A位缺陷对材料析出过程结构与表面形貌的影响
通过甘氨酸-燃烧法制备的三种钙钛矿氧化物材料在经过1100 ℃下烧结2 h后均呈现单一的立方钙钛矿结构,无其它杂相生成,主峰衍射强度高,表明材料成相良好.从图1可以看出,在引入A缺位后,PSFR0.1和PSFR0.2的主峰位置相比于PSFR0向低角度偏移,其主要原因为A位阳离子的缺失导致氧空位形成,从而使晶胞体积增大.在ABO3钙钛矿结构的PSFR0中,A缺位会导致氧空位产生以维持材料的电中性,从而引起(Fe/Ru)O6八面体单元中的B位过渡金属元素价态降低,离子半径增加.Pr0.5Sr0.5FeO3-δ基钙钛矿具有较宽的容差因子范围,在A缺位的量增加时,材料仍保持立方钙钛矿材料,具有较高的结构适应性.
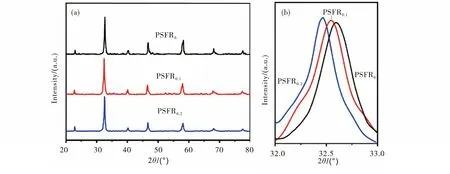
图1 PSFR0,PSFR0.1及PSFR0.2粉体的XRD图谱(a)粉体的XRD图谱;(b)主峰对比Fig.1 XRD patterns of PSFR0,PSFR0.1,PSFR0.2 powders (a) XRD patterns of powders;(b) the comparison of the main peaks
经在加湿H2下还原8 h后,不同A缺位程度的Pr0.5Sr0.5FeO3-δ基钙钛矿的XRD图谱及形貌如图2所示.从图2可见:PSFR0,PSFR0.1和PSFR0.2均发生相转变,钙钛矿主体结构由原来的ABO3立方钙钛矿相转变为RP层状相;于44.23 °处可观察到金属峰,说明在还原过程中发生了金属NPs的析出;经还原后PSFR0的主峰旁边出现了少量的杂质峰,而PSFR0.1与PSFR0.2中没有检测到明显的杂相,说明A缺位的引入可以抑制还原过程中AO相(如Pr2O3和PrO)的形成,在一定程度上保持了RP层状钙钛矿的稳定性.
从图2还可以看出:PSFR0,PSFR0.1及PSFR0.2的表面均生成了分布均匀的Fe-Ru合金的NPs,颗粒的尺寸随着A缺位程度增加而增大,在经过加湿H2气氛下还原8 h后PSFR0,PSFR0.1及PSFR0.2表面的NPs平均尺寸分别为20~40 nm,25~45 nm和45~75 nm.这主要是因为在相同的还原气氛及温度下,A缺位促进了过渡金属元素的析出,使得更多的Fe-Ru金属被还原出来.
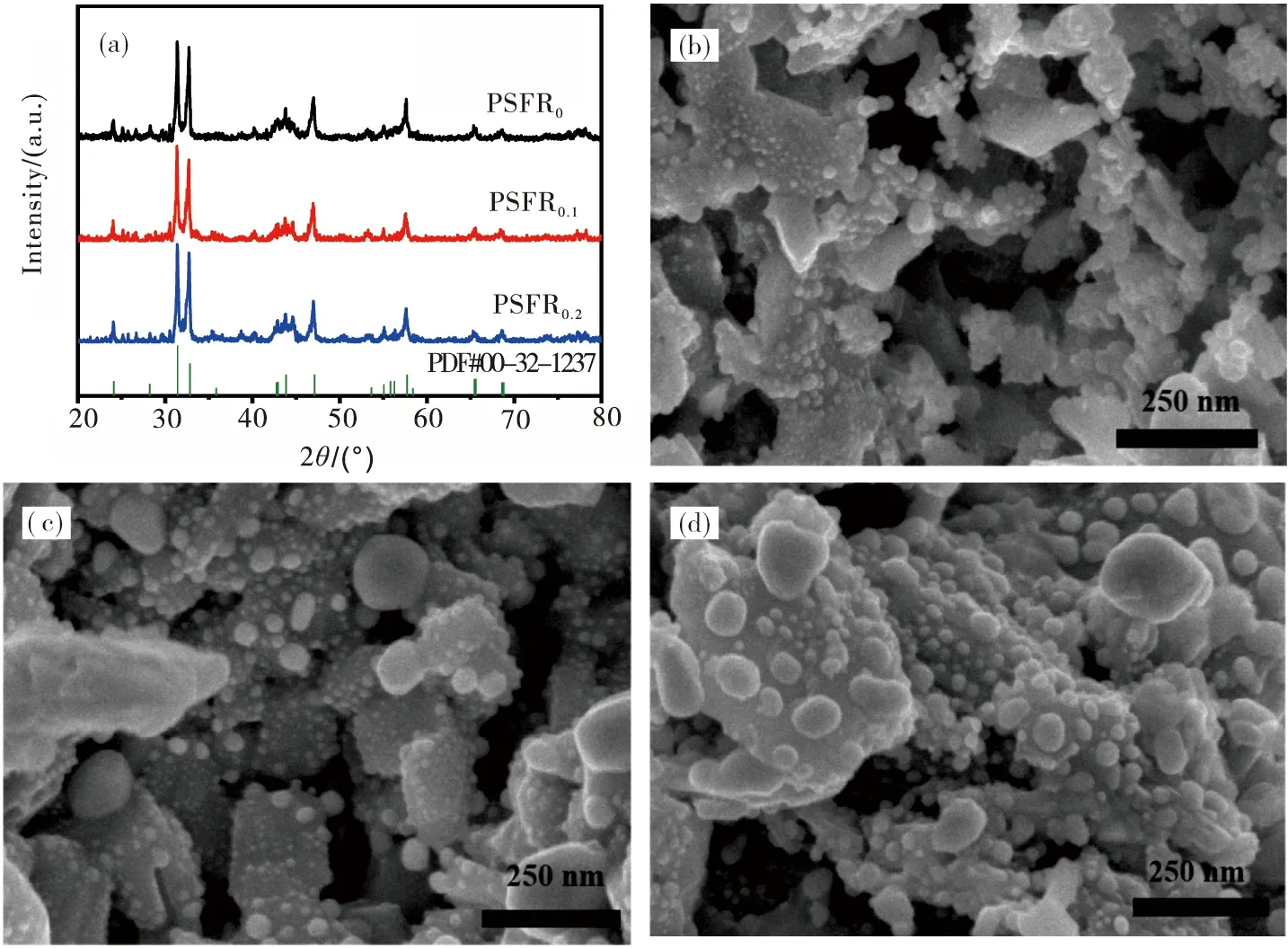
图2 粉体的XRD图谱及微观形貌 (a)XRD图谱;(b)PSFR0粉体形貌;(c)PSFR0.1粉体形貌;(d)PSFR0.2粉体形貌Fig.2 The XRD patterns and the microstructure of the powders (a) XRD patterns;(b) the microstructure of the PSFR0;(c) the microstructure of the PSFR0.1;(d) the microstructure of the PSFR0.2
2.2 对称电池极化阻抗测试
通过对称电池交流阻抗谱,对阳极电化学极化过程进行定量表征.以LSGM为支撑电解质,PSERx-GDC为电极,制备了对称电池.在加湿H2气氛下,测试了不同温度下PSFR0-GDC,PSFR0.1-GDC及PSFR0.2-GDC三种阳极的极化阻抗.为了更直观地对比材料的极化行为,扣除测试所得的欧姆电阻.由于对称电池阴阳极均为同一材料,因此可认为阴极与阳极的极化阻抗相等,即PSFR0-GDC,PSFR0.1-GDC和PSFR0.2-GDC的极化电阻值为半电池总阻抗的1/2.
图3为扣除欧姆电阻后的电极极化阻抗图.从图3(a)~图3(c)可见,三种电极的极化电阻(Rp)均随着温度降低而增加,且主要变化表现在中频和低频段的两个响应;在相同测试条件下,随着A缺位的增加,阳极的极化电阻(Rp)先减小后增加.从图3(d)可以看到,在800 ℃和加湿H2条件下,PSFR0-GDC,PSFR0.1-GDC及PSFR0.2-GDC的电极极化阻抗分别为0.151,0.097和0.205 Ω·cm2,表明引入少量的A缺位可以改善PSFR材料在B位金属析出后的性能.
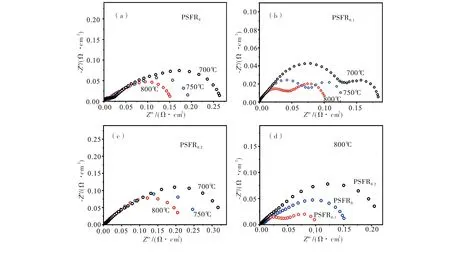
图3 对称电池在加湿H2和不同温度下的交流阻抗谱(a)PSFR0-GDC;(b)PSFR0.1-GDC;(c)PSFR0.2-GDC;(d)800 ℃下三种阳极的电化学极化谱的比较Fig.3 Nyquist plots of EIS data tested with symmetrical cells in humidified H2(a) PSFR0-GDC;(b) PSFR0.1-GDC;(c) PSFR0.2-GDC;(d) the comparison of EIS results of three anodes at 800 ℃
2.3 电池性能及稳定性
图4为以PSFR0-GDC,PSFR0.1-GDC和PSFR0.2-GDC为阳极的LSGM电解质支撑SOFC在加湿H2燃料下测得的电池性能.从图4(a)可见:在800 ℃下,PSFR0-GDC,PSFR0.1-GDC及PSFR0.2-GDC单电池的最大功率密度分别达到了0.7,0.747和0.631 W/cm2;PSFR0.1-GDC单电池表现出最好的输出性能,其在750和700 ℃下的输出功率密度分别为0.523和0.378 W·cm-2,均高于其它两种阳极.从图4(b)~图4(f)交流阻抗谱可看出:在不同温度下,三个电池的欧姆阻抗基本相同,随着温度下降各电池的极化阻抗也随着增大,在800 ℃下PSFR0-GDC,PSFR0.1-GDC及PSFR0.2-GDC单电池的极化阻抗分别为0.098,0.087和0.102 Ω·cm2.
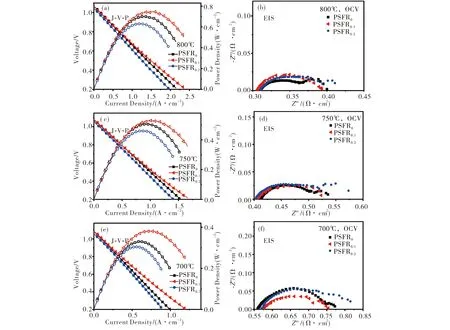
图4 单电池以H2为燃料时的性能输出测试曲线(J-V-P)和对应的电化学阻抗谱(EIS)(a)800 ℃,J-V-P;(b)800 ℃,EIS;(c)750 ℃,J-V-P;(d)750 ℃,EIS;(e)700 ℃,J-V-P;(f)700 ℃,EISFig.4 The performance output test curve (J-V-P) and corresponding electrochemical impedance spectra (EIS) for of single cells with H2 as fuel
单电池的电化学性能测试表明,引入少量的A缺位可以有效地降低材料的极化阻抗,提高输出功率密度.但当A缺位过量时,反而会引起极化阻抗增加,电池性能下降.测试结果表明,PSFRx钙钛矿阳极均表现出较好的功率输出.
以PSFR0-GDC,PSFR0.1-GDC和PSFR0.2-GDC为阳极的LSGM电解质支撑SOFC,在加湿C3H8-N2混合气(C3H8与N2体积比为1∶1)作为燃料气,测试电池性能.图5为电池的J-V-P曲线及电化学交流阻抗谱.从图5可见:在800 ℃下,PSFR0-GDC,PSFR0.1-GDC及PSFR0.2-GDC单电池的最大功率密度分别为0.538,0.528和0.421 W/cm2,表明PSFRx材料对碳氢燃料具有良好的催化性能;PSFR0-GDC和PSFR0.1-GDC单电池在不同温度下的功率密度、欧姆电阻与极化电阻相差不大,但PSFR0.2-GDC单电池的输出性能下降明显.
从图5交流阻抗谱图可以看出,PSFR0.2-GDC单电池的在不同温度下的极化阻抗明显大于PSFR0-GDC和PSFR0.1-GDC单电池,表明PSFR0.2-GDC单电池催化过程中的电荷转移与气体扩散步骤速率远低于其它两者,其性能衰减的主要原因为A缺位程度增加,导致在还原过程中B位金属的过量析出,金属合金NPs生长使得比表面积下降,导致性能衰减.

图5 单电池以C3H8为燃料时的性能输出测试曲线(J-V-P)和对应的电化学阻抗谱(EIS)(a)PSFR0-GDC,J-V-P;(b)PSFR0-GDC,EIS;(c)PSFR0.1-GDC,J-V-P;(d)PSFR0.1-GDC,EIS;(e)PSFR0.2-GDC,J-V-P;(f)PSFR0.2-GDC,EISFig.5 The performance output test curve (J-V-P) and corresponding electrochemical impedance spectra (EIS) for of single cells with C3H8 as fuel
以PSFR0-GDC,PSFR0.1-GDC及PSFR0.2-GDC为阳极的单电池,在750 ℃及加湿C3H8-N2混合气下对单电池进行恒电流放电测试,其中施加电流密度为0.15 A/cm2.图6为单电池的放电曲线.
从图6可以看出,PSFR0-GDC单电池的开始工作电压为0.82 V,经过短时间的衰减后稳定在0.6 V左右,在后续的45 h测试过程中均没有发生进一步的衰减.这说明开始测试后的衰减,主要归因于材料的不稳定性.PSFR0在还原过程中容易随着B位金属的析出而生成杂相Pr2O3和SrO,这些杂相不参与电化学反应步骤,但会阻碍电子与离子的传输,导致电化学性能的衰减.从图6还可见,PSFR0.1-GDC单电池在一开始的输出性能虽然不及PSFR0-GDC单电池,但其可稳定运行超过50 h而没有发生性能的衰减现象,表现出良好的稳定性.这说明少量A缺位的引入,可在促进B位金属析出的同时还能维持还原后材料的稳定性,减少杂相的生成.
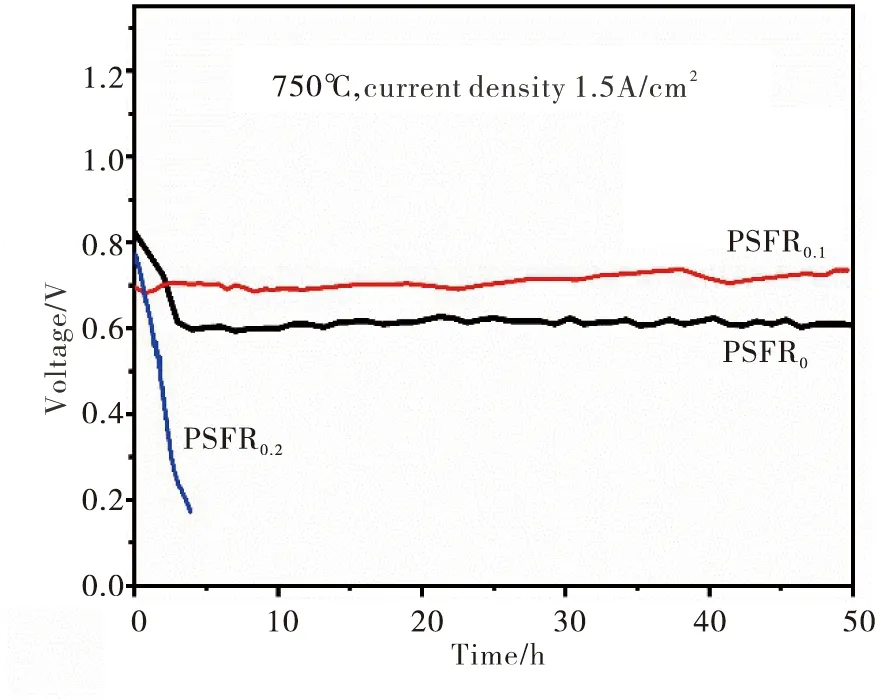
图6 单电池以C3H8为燃料时的恒电流放电曲线Fig.6 Constant current discharge curve of the single cells with C3H8 as fuel
PSFR0.2-GDC单电池在加湿C3H8-N2混合气下性能迅速衰减,这是由于过量的A缺位,会导致在长时间运行过程中B位金属逐渐过量析出而导致NPs长大团聚,减少了反应活性位点而导致材料性能的衰减.
3 结 论
以LSGM为电解质,制备并测试了不同A缺位的钙钛矿阳极材料PSFRx(x=0,0.1,0.2).研究发现,引入少量的A缺位后,可以抑制材料在还原过程中的杂相生成,保证了B位金属析出后的A2BO4层状相的稳定性,改善了材料的电导率及电化学催化性能.同时,A缺位的引入也会促进B位金属的析出,随着A缺位的增加,还原后析出的NPs粒径明显增大.当A缺位过量时,B位金属在还原过程中容易过量析出而导致晶粒长大及团聚,这会极大地降低表面NPs的比表面积,不利于燃料的电化学催化. PSFR0.1表现出最好的电化学性能,其在800 ℃下,以加湿H2及C3H8-N2混合气为燃料时的最大功率密度分别为0.747和0.528 W/cm2,并在丙烷燃料下实现了长期稳定运行.
猜你喜欢
中国粉体技术(2022年2期)2022-03-19
甘肃教育(2021年10期)2021-11-02
粉末冶金技术(2021年1期)2021-03-29
粉末冶金技术(2021年1期)2021-03-29
民主与法制(2020年16期)2020-08-24
传媒评论(2018年8期)2018-11-10
物理学进展(2017年1期)2017-02-23
云南师范大学学报(自然科学版)(2015年5期)2015-12-26
太阳能(2015年4期)2015-02-28
太阳能(2015年2期)2015-02-28
