
儿童肝豆状核变性延误诊断1例*
2021-06-30 01:28吴晓明
中西医结合肝病杂志 2021年6期
吴晓明 何 强 陈 芳 李 歆 莫 鑫 侯 月
国家儿童医学中心首都医科大学附属北京儿童医院中医科 (北京,100045)
1 病历资料
患儿,男,2005年出生,主因“双下肢皮疹12 d,尿检异常9 d”于2015年6月26日住院治疗。患儿入院前12 d无明显诱因出现双下肢红色皮疹,伴瘙痒,无腹痛及关节肿痛,无呕吐、便血,无发热、流涕、咳嗽等。于保定市第二中心医院就诊,查血常规:WBC 9.48×109/L,NEUT% 66.0%,LYMPH% 33.8%,RBC 4.32×1012/L,Hb 122g/L,PLT 303×109/L;尿常规:PRO ++,RBC未见;血生化:ALT 56 U/L,AST 32 U/L,余正常。给予头孢类抗生素(具体不详)口服,病情无好转。入院前7 d出现双侧踝关节肿胀触痛,活动受限,于我院门诊就诊,查血常规:WBC 10.40×109/L,NEUT% 60%,LYMPH% 30.8%,RBC 4.43×1012/L,Hb 129 g/L,PLT 222×109/L,CRP<8 mg/L,诊断:过敏性紫癜,治疗予维生素C、氯雷他啶、头孢丙烯、青紫合剂口服,病情无好转,为求进一步治疗,以“过敏性紫癜、肝功能损害”收入院。家长诉患儿既往体健,否认肝炎、结核等传染病接触史及食物、药物过敏史,父母非近亲结婚,患儿哥哥体健,否认家族遗传病史。入院查体:T 36.8℃,R 22次/min,HR 88次/min,BP 120/75 mmHg,体重25 kg,神清,精神反应可,神情正常,查体合作,皮肤无色素沉着及脱失,周身皮肤黏膜及巩膜无黄染,无肝掌、蜘蛛痣,全身浅表淋巴结未触及肿大,双下肢皮肤可见散在红色皮疹,大小不等,对称分布,稍高出皮面,压之不退色,双侧眼睑可见轻度水肿,咽部稍充血,双侧扁桃体不大,其余未见异常。入院后查血常规:WBC 18.43×109/L,NEUT% 66.0%,LYMPH% 26.2%,RBC 4.79×1012/L,Hb 141g/L,PLT 505×109/L,CRP<8 mg/L;尿常规:PRO +++,RBC未见;24 h尿蛋白定量:57.44~68.2 mg/(kg·d);肝功能:ALT 56 U/L,AST 30 U/L,γ-GGT 65.5 U/L,ALP 69 U/L,Alb 32 g/L,TBil 12μmol/L,DBil 1.8 μmol/L,IBil 9.8 μmol/L;电解质、肾功能、凝血功能、抗链球菌溶血素O抗体、感染性疾病八项筛查(乙型肝炎五项、丙型肝炎病毒抗体、艾滋病病毒抗体、梅毒螺旋体抗体)、EB病毒检测、巨细胞病毒检测、抗核抗体、抗双链DNA抗体、ENA谱、抗中性粒细胞胞浆抗体均未见异常;腹部常规B超:肝肋下3.3 cm,肝实质回声增强、粗糙,回声欠均匀,脾不大。根据患儿具有典型紫癜皮疹、关节肿痛、大量蛋白尿、肝功能异常的临床表现,结合血小板计数正常以及自身抗体检测阴性,确诊为“过敏性紫癜(皮肤关节型)、过敏性紫癜肾炎(肾病型)、肝功能损害”,家长拒绝行肾脏穿刺活检。治疗予醋酸泼尼松片30 mg,bid(2015年7月7日开始)、卡托普利12.5 mg,bid、阿魏酸哌嗪片0.1 g,tid及碳酸钙颗粒口服,复方甘草酸苷静脉滴入保肝治疗,并于2015年7月8日行第1次环磷酰胺0.5 g冲击治疗。经治疗,患儿入院第3天关节肿痛消失,第8天皮疹基本消退,复查肝功能较前无明显变化,2015年7月9日带药出院。出院后患儿继续服用足量泼尼松8周,然后逐渐减量,并每月定期返院行环磷酰胺0.5 g冲击治疗,至2016年1月环磷酰胺共冲击治疗6次(累积剂量为3 g),同时口服门冬氨酸鸟氨酸3 g,bid,葡醛内酯片100 mg,tid治疗肝功能损害。经治疗,患儿自2016年1月开始尿蛋白转阴,此后未再反复。但患儿2015年7月至2016年7月多次查肝功能始终未恢复正常:ALT波动在42.0~303.5 U/L,AST波动在34.9~145.6 U/L,γ-GGT波动在61.3~190.9 U/L,TBA波动在2.91~21.06 μmol/L,胆红素正常。
为进一步治疗,2016年7月7日因“肝功能损害”再次住院治疗。入院查体:生命体征平稳,周身皮肤黏膜及巩膜无皮疹及黄染,咽稍充血,心肺查体未见异常,腹平软,无压痛及反跳痛,肝肋下2 cm,质偏硬,剑突下未触及肝脏,脾未触及,移动性浊音阴性,四肢关节无肿痛及活动受限,神经系统查体未见异常。入院后查尿常规及尿蛋白定量均正常,泼尼松减量至20 mg,qod口服,复查血常规+CRP大致正常;生化全项:TP 53.7 g/L,Alb 33.1 g/L,ALT 50.9 U/L,AST 38 U/L,γ-GGT 52.9 U/L,TBil 21.07 μmol/L,IBil 17.86 μmol/L,TBA 14.98 μmol/L,余大致正常;凝血功能正常;自身免疫性肝病检测阴性;复查腹部常规超声:目前肝肋下2.1 cm,肝内多发小片状低回声;肝胆超声:随机选取肝脏多点测量肝脏弹性,分别为17.7、15.5、17.6、13.1、14.2、15.8、17.4、16.5、18.5、13.4 kPa,肝脏弹性平均值约15.97 kPa。头部MR平扫未见异常。眼科会诊:裂隙灯下K-F环不明显;多次复查铜蓝蛋白均<20 mg/L;尿铜2 003 μg/24 h;基因检测(北京迈基诺基因科技有限责任公司):ATP7B基因有2个杂合突变:c.2122-1G>T 和c.3028A>G。根据儿童肝豆状核变性(HLD)的诊断标准[1],本患儿血清铜蓝蛋白<100 mg/L(2分),尿铜排泄量>80 μg/24 h(2分),单染色体检测到突变(1分),Ferenci评分>4分,故诊断HLD。治疗方案为:低铜饮食;青霉胺开始为125 mg/d口服,逐渐加量至437.5 mg/d口服,待病情改善后逐渐减量,疗程共为2年半;硫酸锌片125 mg,tid,葡醛内酯片100 mg,tid口服至今。治疗1年左右肝功能恢复正常,肝脏超声:肝实质回声粗糙,未见明显低回声灶,肝脏弹性平均值降至6.16 kPa。治疗2年半时尿铜降至42 μg/24 h。治疗3年时腹部超声显示肝脏恢复正常。
2 讨论
HLD又称Wlison病,是一种以铜代谢障碍为主的单基因遗传病,其发病率在全球范围内为1/3万~1/10万,中国的发病率高于西方国家[2]。发病多为儿童和青少年,40岁以上发病者为晚发型,临床上非常少见[3]。位于13号染色体(13q14.3)上的ATP7B基因突变导致铜转运P型ATP酶的结构和功能破坏,引起铜蓝蛋白合成障碍和胆汁中铜排泄减少,使铜在肝、脑、肾、角膜、骨关节、肌肉等多种组织中沉积过多,进而出现相应病变和损害。ATP7B基因突变类型多变且复杂,文献报道致病性变异已超过500种,绝大多数为复合杂合突变,基因突变的差异是导致患者起病年龄不同、临床表现多样、进展快慢不一的主要原因[4]。HLD是少数可治的遗传病之一,早期诊断并尽早启动终身低铜饮食和排铜治疗可使患者不发病或有效缓解病情,从而获得良好的生存质量及与正常人近似的生存期[5]。若未经有效诊疗,重要脏器将发生不可逆损害,预后不佳。
儿童HLD的临床诊断主要依赖于临床表现、血清铜蓝蛋白、24 h尿铜定量、角膜K-F环、肝铜和基因分析[6]。①临床表现:儿童的平均起病年龄约为9岁,临床分型主要有肝型、脑型、肝脑型及其他类型(骨关节症状、肾损害、溶血性贫血),其中肝脏症状出现早,从偶然发现的肝酶异常到肝硬化、急性肝衰竭,临床表现差异很大[7,8]。HLD典型病理特征之一是肝实质弥漫性损伤、肝纤维化,研究显示健康对照组、单纯肝型HLD组、肝脑型HLD组的肝脏弹性平均值分别为(5.14±0.83)、(14.58±7.44)、(20.69±10.07)kPa(P<0.05)[9]。本患儿10岁,ALT、γ-GGT轻度升高,无神经系统的症状和体征,超声显示肝脏肿大,肝实质回声增强、粗糙,肝内多发小片状低回声,肝脏弹性平均值约15.97 kPa,单纯肝型HLD组的肝硬度较为接近,应警惕HLD可能。②血清铜蓝蛋白:铜蓝蛋白的正常值范围是210~530 mg/L,<100 mg/L是临床诊断HLD的重要依据之一[5]。本患儿铜蓝蛋白<20 mg/L,但由于肾病综合征患者铜蓝蛋白也可能呈低水平,患儿在尿蛋白转阴后的3年内多次复查铜蓝蛋白均<20 mg/L,高度提示HLD。③24 h尿铜定量:诊断标准以>100 μg/24 h为阳性,但由于儿童的身高、体重、铜储量均不及成人,按此标准可能误诊,2008年美国推出的HLD指南把尿铜>40 μg/24 h作为筛查对象,我国研究显示24 h尿铜52 μg界值与100 μg界值相比,可显著提高儿童HLD诊断的敏感度和准确性[10],本患儿尿铜为2 003 μg/24 h,明显升高,经排铜治疗2年半后逐渐降至正常,支持HLD。④角膜K-F环:文献报道几乎所有神经系统受累的HLD患者均存在角膜K-F环,然而只有50%~60%的肝型患者有这一表现,儿童K-F环更加少见[4]。我国一项儿童HLD回顾性分析显示神经型患儿K-F环阳性检出率为100%,非神经型患儿K-F环阳性检出率为57.6%(P<0.001),K-F环阳性患儿年龄为(11.8±2.7)岁,阴性患儿年龄为(7.6±3.7)岁(P<0.001)[11]。本患儿10岁,以肝脏病变为主,无神经系统受累证据,未见角膜K-F环亦不能除外HLD。⑤肝铜:肝铜含量是诊断该病的“金标准”,其临界值是250 μg/g干重,正常值为<50 μg/g干重,约有20%(尤其是神经型)患者的肝铜含量<50 μg/g干重。2005年国外研究者将诊断阈值修正为75 μg/g,诊断敏感度提高13%。我国科研人员则通过对178例HLD患者及513例其他肝病患者行肝活检发现,肝铜含量为209 μg/g干重是诊断HLD的最佳临界值,其敏感性和特异性高达99.4%和96.1%[12]。肝铜测定仅用于基因检测不能确诊或无法行基因检测时[6],本例患儿未行肝活检。⑥基因分析:基因检查的特异性为100%,敏感性为60%~85%[13],该样本分析到ATP7B基因有2个杂合突变(见图1):①c.2122-1G>T(编码区第2122-1号核苷酸由鸟嘌呤变异为胸腺嘧啶),导致氨基酸改变splicing(剪接突变),该变异不属于多态性位点,在人群中发生频率极低,在HGMD专业版数据库中未见报道,经家系验证分析,受检人之父该位点无变异,受检人之母该位点杂合变异;②c.3028A>G(编码区第3028号核苷酸由腺嘌呤变异为鸟嘌呤),导致氨基酸改变p.K1010E(第1010号氨基酸由赖氨酸变异为谷氨酸),为错义突变,该变异不属于多态性位点,在人群中发生频率极低,在HGMD专业版数据库中未见报道,经家系验证分析,受检人之父该位点杂合变异,受检人之母该位点无变异。结合本例患儿的临床表现及其他检测结果综合分析,ATP7B基因的复合杂合突变可能是该患儿致病的分子机制。
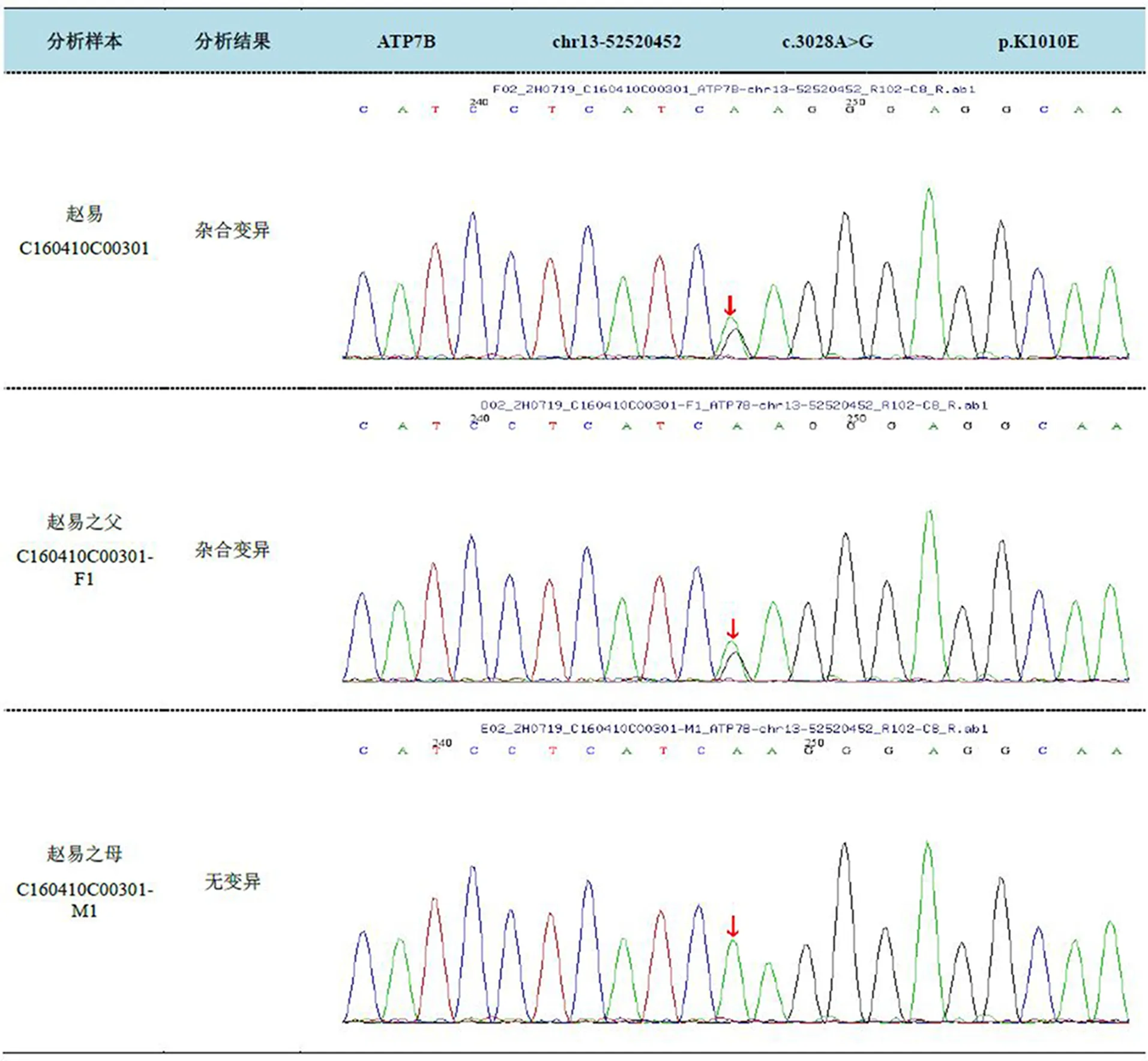
图1 家系分析结果展示
HLD的首发表现多种多样,极易误诊、漏诊。本患儿在发现肝功能损害1年后确诊为HLD,分析本患儿延误诊断的原因:文献报道少数过敏性紫癜患者可同时伴有肝脏转氨酶轻度升高,一般持续2~4周,经保肝治疗以及过敏性紫癜病情好转后肝功能可完全恢复正常,但具体发病机制不详,也可能与前期感染或自服肝毒性药物有关[14]。本患儿因“过敏性紫癜”就诊时发现ALT轻度升高,予保肝治疗,此后因“过敏性紫癜肾炎(肾病型)”开始行环磷酰胺冲击治疗(每月1次,共6次),此时肝功损害不能除外药物性肝损伤的可能[15],继续保肝治疗,待环磷酰胺停药及保肝治疗半年后肝功能仍未恢复正常,随后进行病因筛查确诊为HLD。因此今后临床上应遵循欧洲儿童胃肠病、肝病及营养学会的建议,对1岁以上出现无症状性转氨酶升高、肝硬化合并肝脾肿大或腹水、急性肝衰竭等表现的患儿均应排除HLD[6]。
此外,既往有肝相关性IgA肾病的文献报道:IgA肾病可见于成人肝硬化和门静脉高压症患者[16];王倩等[17]曾报告1例8岁HLD患儿以血尿、水肿为首发表现,肾活检提示为典型的IgA肾病;Alghamdi等[18]报告1例14岁患有隐匿性肝硬化和门静脉高压的男孩发生血尿和蛋白尿,肾脏病理提示为IgA肾病的组织学改变;Singhal等[19]报告1例自身免疫性肝病患儿出现血尿、蛋白尿,并逐渐进展至慢性肾脏病Ⅲ期,肾活检显示肾脏系膜区有IgA免疫复合物沉积。人们认为慢性肝病(包括HLD)可引发IgA肾病[20],认为其发病机制可能是慢性肝病时肝脏Kupffer细胞对循环免疫复合物的清除能力下降,或者门脉高压造成免疫复合物无法被肝脏清除,使免疫复合物进入循环系统并沉积于肾脏,从而引发IgA肾病[18,21],有报道称改善门脉高压等肝脏疾病后可以减轻IgA肾病的血尿、蛋白尿[22]。但也有专家提出上述两种疾病的关联只是一种推测,并无确切证据表明IgA肾病是继发于慢性肝脏疾病,很可能是发生于同一患者的两个单独的疾病[23]。由于过敏性紫癜与IgA肾病具有共同的发病机制,即糖基化异常的IgA1分子介导的免疫学异常,有学者提出IgA肾病是无皮疹的过敏性紫癜肾炎[24],因此笔者认为,今后需要更进一步深入研究慢性肝病(包括HLD)与过敏性紫癜肾炎的发病之间是否存在关联性。
猜你喜欢
中国中医药信息杂志(2022年3期)2022-03-16
家庭医学(2020年11期)2020-12-28
恋爱婚姻家庭(2020年36期)2020-12-25
婚育与健康(2017年9期)2017-11-04
家庭医学·下半月(2017年4期)2017-06-02
家庭·育儿(2009年9期)2009-09-22
中国社区医师(2009年12期)2009-08-12
浙江中医杂志(2004年4期)2004-04-26
祝您健康(1994年8期)1994-12-30
