
44例假性醛固酮减少症Ⅰ型临床分析并文献复习
2021-06-23 06:50:32綦奕颖吴道奇
儿科药学杂志 2021年6期
綦奕颖,吴道奇
(重庆医科大学附属儿童医院,重庆 400014)
假性醛固酮减少症(pseudohypoaldosteronism,PHA),又称醛固酮不敏感综合征或假性低醛固酮血症,是一种发生于新生儿期及婴儿期的罕见失盐综合征,由Cheek D B和Perry J W于1958年首次报道,又称Cheek-Perry综合征[1]。PHA分为Ⅰ、Ⅱ、Ⅲ型,PHAⅠ又可分为多脏器缺陷型和单纯肾型[2]。单纯肾型PHAⅠ为常染色体显性遗传病,其肾小管盐皮质激素受体(MCR)对醛固酮不敏感,钠盐丢失,病变局限于肾脏,表现为脱水、低钠血症和高钾血症等电解质紊乱,病情较轻者可随年龄增长自行缓解[3-4]。多脏器型PHAⅠ系常染色体隐性遗传病,其发病机制与编码上皮细胞钠离子通道(ENaC)亚基的基因突变有关[5-6],表现为低钠血症、高钾血症、脱水和代谢性酸中毒等,也可合并呼吸道感染、胆汁淤积和皮疹等[7]。PHAⅠ起病年龄较小,临床症状多样化且不典型,极易误诊漏诊,确诊需基因检测。笔者总结分析44例PHAⅠ的临床特点,为临床早期诊断、及时治疗提供参考。
1 资料和方法
回顾性分析重庆医科大学附属儿童医院建院至2018年6月从住院患儿病案中搜集的住院确诊为PHAⅠ的病例4例,同时从PubMed、CNKI、万方数据库、中文科技期刊数据库和中国生物医学文献数据库中检索明确报道的PHAⅠ病例40例(检索时间从建库至2018年6月,语种限中文和英文,排除重复报道的病例)[8-31],合并后共44例患儿(通过相关临床表现及基因检测确诊为PHAⅠ),分析其临床表现、发病特点及诊疗经过等。
2 结果
2.1 发病特点及临床表现
44例患儿发病年龄均较小(0~2.6个月),平均为10.2 d,高峰在生后1周左右。发病时均有不同程度的多种临床表现,其中电解质紊乱(高血钾、低血钠)(100%)、多尿(79.5%)、脱水(65.9%)、神萎/反应差(31.8%)、体质量不增/生长受限(29.5%)、皮疹/色素沉着(29.5%)、腹泻(27.3%)、代谢性酸中毒(22.7%)、拒食/纳差(20.5%)、吐奶(15.9%)、心律失常(13.6%)、反复呼吸道感染(11.4%)、烦躁/易哭吵(6.8%)、发热(4.5%)、抽搐(4.5%)。
2.2 实验室检查特点
44例患儿实验室检查示均有高血钾(K+最高为10.49 mmol/L)、低血钠(Na+最低为114.40 mmol/L)的显著特点;血气分析示10例有不同程度的代谢性酸中毒;尿、大便、汗腺和唾液中钠钾比值增大;肾小球滤过率正常;醛固酮、肾素活性升高;基因检测突变分别为SCNN1A、SCNN1B、SCNN1G及NR3C2。
2.3 基因突变类型分布
5例单纯肾型PHAⅠ中有4例未提供相关基因检测结果,39例多脏器型PHAⅠ中有1例未发现基因突变,共计39例有相关基因突变检测结果,包括SCNN1A突变29例、SCNN1B突变6例、SCNN1G突变3例、NR3C2突变1例(单纯肾型)。
2.4 误诊情况
44例患儿病初均有误诊,误诊率较高。由于PHAⅠ临床表现复杂多样,临床上易被误诊为先天性肾上腺皮质增生症(CAH)、先天性肾上腺皮质功能减退症、醛固酮减少症、新生儿缺氧缺血性脑病(HIE)、继发型PHA、Ⅳ型肾小管酸中毒、消化道畸形、18羟化酶缺乏等疾病。PHAⅠ与常见误诊相关疾病的鉴别诊断见表1[8]。
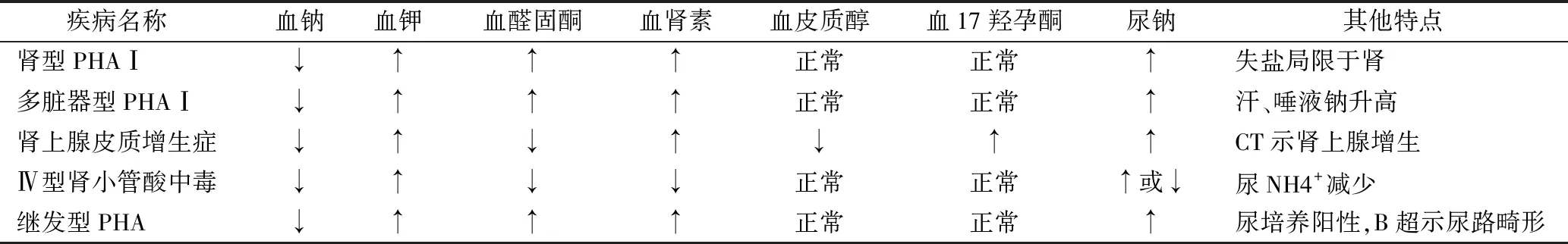
表1 PHAⅠ与常见误诊疾病的鉴别诊断
2.5 治疗及转归
PHAⅠ患儿存在住院时间较长、需多次抢救、腹膜透析、血液净化以及因反复呼吸道感染多次住院治疗等问题。尤其多脏器型PHAⅠ病情较重,生后即可出现失盐症状,可发生致死性高钾血症,易发生下呼吸道感染,表现为反复发作的呼吸困难、发热、呼吸急促及三凹征,需补充大剂量钾盐以代偿严重的多脏器失盐。多脏器型PHAⅠ在新生儿期起病,急性期面临的问题主要是电解质紊乱,特别应警惕高血钾,早期累及呼吸系统等肾外系统的表现尚不明显,抢救不及时,可能危及生命。远期随访发现,肾外表现可能逐一显现,影响患儿生活质量及增加住院概率。本研究中,5例单纯肾型PHAⅠ患儿病情均较轻,经治疗后预后均较好。其中,4例未提供相关基因检测结果,1例为NR3C2突变;5例患儿经高钠饮食后,低钠、高钾和酸中毒得到纠正,经低钾饮食等对症治疗后病情好转。39例多脏器型PHAⅠ患儿中,5例由于抢救或治疗不及时,因高钾、酸中毒、脱水无法纠正或继发感染等多种危险因素出现导致多器官衰竭而死亡,基因检测均提示为SCNN1A突变(纯合突变3例,杂合突变2例);3例经过多次纠酸、补钠、腹膜透析、血液净化等抢救后,达到临床痊愈,均为SCNN1A突变(纯合突变2例,杂合突变1例);14例经长期补钠及口服降钾树脂后电解质保持平稳,包括无基因突变1例,SCNN1A突变8例,SCNN1B突变2例,SCNN1G突变 3例;17例在长期补钠及口服降钾树脂情况下仍间断出现失盐症状、电解质不稳定,并伴有呼吸道感染或皮疹等症状,包括SCNN1A突变13例,SCNN1B突变4例。
3 讨论
PHAⅠ是临床上罕见的一种失盐综合征,为常染色体隐性或显性遗传。多在新生儿期发病,可于生后数小时出现症状,存在严重电解质紊乱,主要表现为致死性高血钾、低血钠,以反复呕吐、腹泻、渴感减退或消失、多尿、脱水、酸中毒及生长发育落后为主要症状,有的则在限盐或应用醛固酮拮抗剂后才出现症状。其中,累及多脏器的为常染色体隐性遗传的多脏器型,仅累及肾脏的为常染色体显性遗传的单纯肾型。
3.1 PHAⅠ病因及发病机制
PHAⅠ的病因为靶器官(肾小管、唾液腺、汗腺和结肠)上的醛固酮受体缺乏或醛固酮与其受体结合减少或完全不能结合,分子生物学及分子生物化学的进一步研究发现,PHA的病因学基础是由基因决定的细胞膜上钠通道功能障碍。其发病机制:人体内水钠代谢平衡受醛固酮的调控。醛固酮的作用通过醛固酮受体(MR)传导。MR由NR3C2基因编码。NR3C2基因定位染色体4q31.1,含有10个外显子。MR信号通路通过激活阿米洛利敏感的ENaC储存细胞内钠[9]。ENaC含有α、β和γ三个亚基,只有当这三个亚基同时表达才可获得最大的钠通透性[8],分别由SCNN1A(定位12P13染色体)、SCNN1B及SCNN1G(定位16p12)三个基因编码,与常染色体隐性遗传病相关,这些基因突变不仅可影响肾脏,还可影响唾液腺、结肠、汗腺和呼吸道[9-10]。因此,单纯肾型PHAⅠ是由于NRA3C2突变,导致MR数量或功能缺陷;多脏器型PHAⅠ是由于ENaC的α或β亚基基因失活性突变,导致存在ENaC的组织器官钠离子转运缺陷。由于遗传缺陷、靶组织皮质激素缺陷或细胞功能障碍损害了醛固酮的作用,导致盐皮质激素受体减少或缺乏。由于醛固酮作用的减弱,远端肾单位对钠的主动重吸收障碍,致钠从尿中大量流失,氧和水的重吸收明显减少,最终导致失盐表现。另外,由于钠的主动重吸收障碍致肾小管腔内负电位差降低,远端肾单位主动分泌H+和被动排泌K+功能发生障碍,导致尿酸化形成碱性尿和钾排出障碍引起高钾血症、酸中毒。
3.2 PHAⅠ临床表现分析
通过对本研究中44例PHAⅠ进行分析,可将PHAⅠ的临床表现总结为高血钾、低血钠、代谢性酸中毒等,可伴有呕吐、腹泻、脱水、渴感减退或消失等;如未经正规治疗及时纠正电解质紊乱,可出现嗜睡、低体温、循环衰竭、喂养困难、体质量不增、生长发育落后等;部分患儿还会出现呼吸系统病变。血液生化检查呈低钠、低氯和高钾,伴或不伴酸中毒,同时存在高血浆肾素活性及高醛固酮血症为PHAⅠ的特征性改变。不同患儿受累的靶器官不尽相同,失盐程度不一,多因高尿钠引起多尿、低渗或等渗性脱水、严重电解质紊乱,如未及时采取有效治疗,多因脱水或继发感染而死亡。
3.2.1 单纯肾型 单纯肾型PHAⅠ发生于新生儿期及婴儿期,随着年龄的增长,病情会有所改善。患儿表现为口渴、多饮、恶心呕吐、脱水、厌食、体质量下降、软弱无力、心律不齐、生长发育停滞等。反复脱水可以引起休克、昏迷。婴儿期后生长发育停滞更明显,易出现低血容量、低血压、类似真性醛固酮缺乏症。幼儿期高钾血症仅表现出呕吐的症状,生长发育停滞可能是此型患儿的唯一体征。年长儿可出现嗜盐的表现,高血钾可引起室上性或室性心动过速等心律失常表现,严重者心脏停搏而导致死亡。
3.2.2 多脏器型 多脏器型PHAⅠ发生于新生儿期及婴儿期,可持续至成人。临床表现与单纯肾型PHAⅠ类似,症状更为严重,生后可迅速出现失盐的表现。合并呼吸、消化系统及皮肤症状,表现为反复呼吸困难、发绀、肺部湿啰音、腹泻、多汗等。
3.3 PHAⅠ的诊断依据
PHAⅠ可通过相关临床表现结合基因检测确诊。相关临床表现:(1)低钠、高钾血症,低氯等,部分患儿可有酸中毒;(2)大小便、汗腺和唾液中排钠增多,唾液呈咸味,尿中醛固酮排量增大;(3)生长落后、体质量减轻;(4)喂食困难,尤其是伴有肌肉痉挛和肌张力减退者;(5)反复发作性急性呼吸困难、咳嗽和哮喘;(6)血浆肾素活性(PRA)和醛固酮醛缩酶(ALD)明显增高;(7)对外源性盐皮质激素治疗无反应。
3.4 PHAⅠ相关鉴别诊断
3.4.1 真性低醛固酮血症 获得性原发性醛固酮缺乏症表现为失盐,多为肾上腺皮质功能减退,血皮质醇和尿17-羟皮质类固醇均降低,血浆醛固酮降低。
3.4.2 失盐综合征 如21-羟化酶缺乏症、18-羟化酶缺乏症,除有失盐表现外,同时有外生殖器发育异常,即女性男性化或男性假性性早熟,血浆肾素活性和醛固酮浓度往往低于正常水平。血促肾上腺皮质激素(ACTH)明显升高而血浆皮质醇明显降低,临床上用皮质醇治疗有效。
3.4.3 肾性失盐性肾炎 肾性失盐性肾炎多有原发病的病因,多为成人起病,患者可表现为低血钠、脱水,但不属于遗传病。
3.4.4 肾小管性酸中毒 肾小管性酸中毒除低血钠外,尚有低血钙、高血氯、低血钾等,可与PHAⅠ鉴别。
3.5 PHAⅠ的治疗方案分析
PHAⅠ的基本治疗为补充氯化钠,纠正酸中毒和高钾血症。绝大多数患者可在2岁左右停止治疗。治疗有效的指标为患者的失盐状态纠正,渴感恢复,生长发育恢复正常,血浆肾素活性和血醛固酮浓度下降或恢复正常。
3.5.1 补钠 补钠量8~50 mmol/(kg·d),达到钠平衡。随着年龄增长,补钠量逐渐减少;大多数患儿2岁时可停止补钠,在饮食中增加氯化钠量。在病情危急时应静脉补充生理盐水或3%高渗盐水。
3.5.2 高钾血症的治疗 血钾过高常引起严重心律不齐或停搏,此时应采取紧急措施进行抢救:(1)静脉滴注的10% GS或生理盐水10 mL/kg,中途加胰岛素0.15~0.20 U/kg,静脉滴注时间>2 h,促使血钾转入细胞内;(2)采用腹膜透析或血液净化;(3)多脏器型PHAⅠ应给予低钾饮食0.6 mmol/(kg·d),限制含钾药物和食物,避免输注库存血;(4)10%葡萄糖酸钙1~2 mL/kg+等量10% GS;(5)5% NaHCO33~5 mL/kg,可重复2~3次;(6)钾离子交换树脂亦有助于降低血钾[11]。
PHAⅠ临床上罕见,其确诊依据只有基因检测,但基因检测时间较长。PHAⅠ起病急,病情重,临床表现多样化,因临床医师认识不够,极易发生误诊及延误治疗的情况。多数患儿因高尿钠引起多尿、低渗或等渗性脱水、严重电解质紊乱,如致死性高血钾等出现病情危重及严重并发症时,很可能因抢救不及时而危及生命。致死性高血钾需通过透析、血液净化等抢救治疗,不仅风险高,也增加了治疗费用。若能通过总结PHAⅠ的临床表现及发病特点,对疾病进行早期预测,尽早进行基因突变的筛查,及时给予有效降钾、补钠、补液、纠酸等对症支持治疗,或可挽救患儿生命。本研究通过对目前已确诊病例的综合分析及总结,对PHAⅠ病例进行学习及归纳,对提高临床医师对疾病的认识及早期识别能力,具有重要临床意义。
猜你喜欢
中国畜禽种业(2021年1期)2021-12-02 15:17:37
现代畜牧科技(2021年8期)2021-10-13 07:22:08
现代畜牧科技(2021年4期)2021-07-21 06:13:26
中国毕业后医学教育(2020年6期)2020-12-06 07:30:04
透析与人工器官(2020年1期)2020-11-16 01:42:28
重庆医学(2019年13期)2019-02-19 02:17:17
兽医导刊(2018年21期)2018-03-18 02:59:58
首都食品与医药(2018年14期)2018-03-16 23:26:16
医药前沿(2018年3期)2018-01-19 06:08:26
首都食品与医药(2017年22期)2017-04-04 07:41:54
