
P-I-N型锡铅钙钛矿太阳电池性能的限制因素及解决策略*
2021-06-18 08:41:32王俪璇李仁杰刘辉王鹏阳石标赵颖张晓丹
物理学报 2021年11期
王俪璇 李仁杰 刘辉 王鹏阳石标 赵颖 张晓丹†
1) (南开大学, 光电子薄膜器件与技术研究所, 太阳能转换中心, 天津 300350)
2) (天津市光电子薄膜器件与技术重点实验室, 天津 300350)
3) (薄膜光电子技术教育部工程研究中心, 天津 300350)
4) (化学科学与工程协同创新中心, 天津 300072)
5) (南开大学可再生能源转换与储存中心, 天津 300072)
锡铅钙钛矿太阳电池已被证明可以用于全钙钛矿叠层太阳电池中, 作为窄带隙底电池进一步提高器件光电转换效率.目前, P-I-N型锡铅钙钛矿太阳电池的最高效率为21.7%, 明显低于铅基钙钛矿太阳电池.本文分析了限制其性能提高的主要因素, 并针对性地总结了近几年研究工作者们提出的有效解决策略, 主要包括: 1)通过添加富锡化合物、强还原剂或含大的有机阳离子的化合物以抑制Sn2+氧化, 减少锡铅钙钛矿材料p型掺杂程度, 降低电池开路电压损耗; 2)通过调控组分、改变钙钛矿薄膜制备方法、溶剂工程或添加含功能性基团的化合物以延缓锡铅钙钛矿薄膜结晶生长速率, 提高薄膜质量; 3)通过选用合适的电子传输层或空穴传输层, 减少能级失配对载流子传输的影响或避免载流子传输层的本身不稳定性对器件的影响.最后, 本文展望了锡铅钙钛矿太阳电池的未来发展, 认为其不仅有望实现高效稳定的单结太阳电池, 而且还可以应用于高效全钙钛矿叠层太阳电池.
1 引 言
自2009年Miyasaka等[1]首次使用钙钛矿作为光吸收层达到3.8%的功率转换效率(power conversion efficiency, PCE)以来, 铅基钙钛矿由于其出色的光学和电学性能引起光伏领域研究工作者们的广泛关注.到2020年, 铅基钙钛矿太阳电池(perovskite solar cells, PSCs)的PCE已达到25.5%[2].尽管铅基钙钛矿太阳电池的光伏性能令人鼓舞, 但单结钙钛矿太阳电池的PCE始终受到Shockley-Queisser (SQ)辐射极限的限制[3].突破SQ辐射极限的一种有效方法是制备由仅吸收高能光子的宽带隙顶电池和主要吸收低能光子的窄带隙底电池组成的叠层太阳电池.目前钙钛矿/硅叠层太阳电池PCE已达到29.15%[2], 但钙钛矿/钙钛矿叠层电池由于缺少高效的窄带隙钙钛矿太阳电池, 实验室最高PCE仅25.6%[4].
已有大量研究证明, 使用Sn2+部分或全部代替Pb2+以制备含锡钙钛矿材料, 带隙可以实现从1.55 eV到1.17 eV的范围内连续调节[5—9].因此,含锡钙钛矿可用作全钙钛矿叠层太阳电池中窄带隙底电池的吸收层材料.并且, Sn2+与Pb2+有相似的离子半径(Sn2+为110 pm, Pb2+为119 pm)及相似的ns2np2电子结构[10—12].因此, 用锡替代铅可以保留原本的钙钛矿结构, 不会引起明显的晶格畸变且能保持与铅基钙钛矿基本相同的光电特性.另外, 从环境友好的角度考虑, Li等[13]的研究结果表明, 铅基钙钛矿中的铅泄漏到地下后可以进入植物体内, 从而进入食物循环, 最终对人体和自然造成伤害.相比较而言, 尽管锡对人类和动物的毒性比铅高, 但在环境中分散后, 钙钛矿中的Sn2+会迅速氧化成稳定的Sn4+化合物, 其低水溶性会降低生物利用度[14].因此, 开发少铅和无铅的含锡钙钛矿太阳电池具有非常重要的研究意义.
目前锡铅(Sn-Pb)钙钛矿太阳电池的最高PCE为21.7%(认证PCE为20.7%)[4], 纯锡钙钛矿太阳电池的最高PCE仅为13.24%[15], 远低于Sn-Pb钙钛矿太阳电池.这是由于相比于纯锡钙钛矿材料, 含有一定比例的Pb2+可增加破坏金属-卤化物-金属键所需的能量, 增加锡空位的形成能从而抑制锡空位的形成, 因此保留一定比例的铅可提高器件的光伏性能及稳定性[16].换而言之, 相比于纯锡钙钛矿太阳电池, Sn-Pb钙钛矿太阳电池具有更大的开发潜力.
但相比于铅基钙钛矿太阳电池, Sn-Pb钙钛矿太阳电池的发展也面临很多挑战.第一, 处于化学元素周期表第六周期的铅原子由于镧系收缩引起的惰性电子对效应, 其6s2的电子很难失去, 而于第五周期的锡原子的5s2电子遇到氧气等强电负性物质时非常不稳定, Sn2+极易被氧化为Sn4+, 即使在极少量氧气的手套箱中也不可完全避免[10,17,18].并且由于Sn2+容易被氧化成Sn4+, 在成膜过程中将从钙钛矿晶格中被排除而留下大量锡空位, 从而会在钙钛矿材料中导致p掺杂, 使载流子寿命缩短, Urbach能量更高, 进而导致开路电压损耗严重, 降低器件性能和可重复性[9,19—23].第二, 由于Sn2+的路易斯酸度高于Pb2+, SnI2与有机碘化物的亲和力更强, 从而更加迅速地形成钙钛矿相, 阻碍了薄膜的均匀生长, 增加了控制Sn-Pb钙钛矿薄膜形貌的难度[24,25].第三, 由于Sn-Pb钙钛矿太阳电池的发展晚于铅基钙钛矿太阳电池, 目前Sn-Pb钙钛矿太阳电池通常使用铅基中的载流子传输层, 但由于锡的引入会导致钙钛矿材料能带位置改变并且Sn-Pb钙钛矿材料本身的不稳定性, 因此原有的载流子传输层会导致Sn-Pb钙钛矿太阳电池界面能级不匹配并且加剧了器件的不稳定性[5,8,9,26,27].以上三方面因素都会严重影响Sn-Pb钙钛矿太阳电池的光伏性能及稳定性.
Wang等[28]在总结近些年Sn-Pb钙钛矿太阳电池的文献报道中, 发现制备N-I-P型结构器件的研究非常少, 并且其最高PCE仅为14.04%[29], 远低于P-I-N型结构器件的性能, 他们把这个现象归因于以下几点: 1) N-I-P型结构器件中, 下层的金属氧化物电子传输层会影响随后沉积的Sn-Pb钙钛矿薄膜结晶质量[8,30]; 2)金属盐(如Li和Co盐)常掺杂在N-I-P结构器件的空穴传输层(例如,spiro-OMeTAD或P3HT)中, 会损坏Sn-Pb钙钛矿薄膜并导致较差的器件性能.为了实现良好的空穴传输性能, 还需要进行氧化工艺[5]; 3)铅基钙钛矿太阳电池中常用的载流子传输层可能与Sn-Pb钙钛矿材料能级不匹配, 导致器件性能不高[26,27].并且, 在全钙钛矿叠层电池中通常采用P-I-N型结构器件.因此本文将着眼于P-I-N型Sn-Pb钙钛矿太阳电池的发展, 为后续Sn-Pb钙钛矿太阳电池性能的快速提升及在全钙钛矿叠层电池中的应用奠定基础.
本文总结了近年P-I-N型Sn-Pb钙钛矿太阳电池的研究进展(图1—图3, 表A1); 第2节分别讨论了目前抑制Sn2+氧化、提高Sn-Pb钙钛矿薄膜结晶质量、选择合适的载流子传输层的主要策略; 最后, 对Sn-Pb钙钛矿太阳电池的发展提出展望.
图1 提高P-I-N型锡铅钙钛矿太阳电池性能的解决策略Fig.1.Solutions to improve the performance of P-I-N type tin lead perovskite solar cells.

图2 钙钛矿材料的能带结构随着金属比例的变化而调整 (a) Ogomi等[5]表征CH3NH3Sn1—xPbxI3能带结构随金属比例的变化; (b) Eperon等[7]通过Tauc plot、PL及第一性原理计算FASnxPb1—xI3带隙随金属比例的变化趋势;(c) Hao等[8]通过紫外吸收光谱表征CH3NH3Sn1—xPbxI3的带隙变化Fig.2.The energy band of Sn-Pb perovskite changed with the metal ratios: (a) Ogomi et al.[5] characterized the CH3NH3Sn1—xPbxI3 energy band structure changed with the metal ratio; (b) Eperon et al.[7] used the Tauc plot, PL and first-principles calculations to obtain the variable trend of HC(NH2)2SnxPb1—xI3(FASnxPb1—xI3) band gap with metal proportions; (c) Hao et al.[8] characterized the band gap changes of CH3NH3Sn1—xPbxI3 by electronic absorption spectra.

图3 不同结构的Sn-Pb钙钛矿太阳电池器件效率进展图, 包括N-I-P型(绿色)和P-I-N型(红色)结构Fig.3.The efficiency progress diagram of Sn-Pb perovskite solar cells in different structures, including N-I-P (green)and P-I-N (red) structures.
2 Sn-Pb钙钛矿太阳电池性能提高的主要方法
2.1 抑制Sn2+氧化
如引言中所讲, 薄膜内Sn2+的氧化会形成大量锡空位, 导致内部缺陷密度增加, 引起严重的载流子复合, 载流子寿命缩短, 开路电压损耗严重,降低器件性能.关于Sn-Pb钙钛矿中Sn2+的氧化机制, Leijtens等[16]结合X射线衍射(X-ray diffraction, XRD)、热重分析(thermogravimetric analysis, TGA)和紫外-可见吸收光谱(UV-Vis), 探究了ASnI3和ASn0.5Pb0.5I3的氧化降解途径及最终产物, 发现纯锡钙钛矿的氧化降解机制如(1)式:
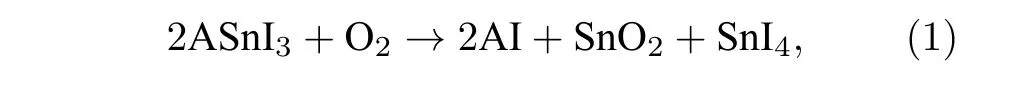
Sn-Pb钙钛矿的氧化机制如(2)式:
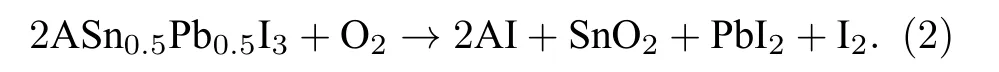
相比于纯锡钙钛矿的氧化分解仅需断裂小部分Sn—I键, Sn-Pb钙钛矿则需要断裂所有的Sn—I键, 并且铅不易被氧化成Pb4+, 不太可能形成PbI4.因此, 如果用一定量的Pb去替代钙钛矿组分中的Sn, 就可以有效抑制钙钛矿的氧化, 即可以通过调控Sn-Pb的比例抑制Sn2+的氧化.
但如果固定钙钛矿中Sn-Pb的比例后, 要实现抑制Sn2+的氧化, 则可能需要结合添加剂的作用.目前能够起到抑制Sn-Pb钙钛矿中Sn2+氧化作用的添加剂主要分为三类: 1) SnF2添加剂, 能够有效补偿Sn2+损失; 2)还原剂, 能够将被氧化的Sn2+还原; 3)添加大的有机阳离子, 形成混合2D-3D异质结构钙钛矿太阳电池.
1) SnF2.SnF2是目前Sn-Pb钙钛矿制备中最常使用的添加剂.由于在Sn-Pb钙钛矿中很容易形成由锡空位引起的固有缺陷, 而富Sn2+的SnX2添加剂可以有效提高锡空位的形成能[31], 因此对于抑制Sn-Pb钙钛矿薄膜中缺陷的形成是非常重要的.SnF2是SnX2系列中被最广泛应用于制备Sn-Pb钙钛矿薄膜的化合物, 这是由于SnX2的键合能会随卤化物离子半径的减小而增加, SnF2是SnX2系列中溶解度最低的化合物.在旋涂过程中随着溶剂蒸发, 不完全溶解的SnF2可以作为非均相成核位点以促进Sn-Pb钙钛矿晶体的生长并实现更均匀覆盖的钙钛矿薄膜[32].除了对薄膜掺杂、形貌产生影响, SnF2也会影响Sn-Pb钙钛矿中的晶相形成、材料稳定性以及能级位置[33].过量的SnF2会引起钙钛矿薄膜中相分离, 如图4(a)所示, 这会破坏薄膜的形貌, 不利于器件的长期稳定性[34,35].为避免SnF2在薄膜中引起的局部偏析, Zong等[36]通过在(FAPbI3)0.7(CsSnI3)0.3钙钛矿前驱体溶液中加入新的路易斯加合物SnF2·3FACl, 使钙钛矿晶粒被均匀地覆盖一层低结晶度且高度连续的SnF2相, 在晶界处有效地抑制了Sn-Pb钙钛矿薄膜中固有的锡空位的形成, 而且还形成了单个钙钛矿晶粒的纳米级封装层以阻挡湿气/氧气进入薄膜.另外SnF2还可以作为化学合成中的强还原剂,可以抑制钙钛矿薄膜的氧化, 减少背景载流子密度[31,33,37].但是Xu等[38]发现仅使用SnF2不能完全抑制前驱体溶液中Sn2+氧化, 因此需结合其他还原剂或寻求还原性更强的还原剂.
2)还原剂.目前很多有效的还原性物质已被应用于锡铅钙钛矿中, 减小Sn2+的氧化程度.研究者们主要选用的是含有烷羟基(—R—OH)、酚羟基(—Ph—OH)、次磷酸根()等还原性基团的化合物或能与Sn(Ⅳ)发生归中反应的Sn(0)物质.Xu等[38]使用抗坏血酸(AA)结合SnF2添加到MA0.5FA0.5Pb0.5Sn0.5I3前驱体溶液中, 利用AA中的羟基不仅可以作为共轭的路易斯碱与作为路易斯酸的SnI2形成中间体络合物改变钙钛矿的结晶过程, 还可以延缓Sn2+的氧化.这种方法将钙钛矿膜内光生载流子寿命提高至182.7 ns, 可以实现14.01%的PCE, 优于使用常规SnF2添加剂处理的12.18%的器件PCE.Zhu等[39]利用伽伐尼置换反应(Galvanic replacement reaction, GDR),将极细(—100目)锡金属粉末与铅基钙钛矿前驱体溶液混合搅拌, 图4(b)中, 制备的MAPb0.4Sn0.6I3及(FAPb0.6Sn0.4I3)0.85(MAPb0.6Sn0.4Br3)0.15分别表现出15.85%及18.21%的PCE.Lin等[18]发现Sn2+的氧化问题主要发生在SnI2固体及锡铅钙钛矿前体溶液中, 因此他们利用金属锡的还原性, 向MA0.3FA0.7Pb0.5Sn0.5I3钙钛矿添加金属锡粉末与SnF2(相比于SnI2为10 mol%), 如图4(c)所示, 利用归中反应(Sn+Sn4+→Sn2+)将Sn2+的氧化产物Sn4+还原为Sn2+, 大大减少Sn空位且载流子扩散长度增加至3 μm.对于1.22 eV的Sn-Pb钙钛矿太阳电池获得21.1%的PCE, 应用在小面积(0.049 cm2)全钙钛矿叠层太阳电池获得24.8%的PCE.最近, 他们又考虑到前体溶液中所使用的溶剂对结晶过程中Sn2+氧化的影响[40], 于是在前述工作的基础上使用纺织工业上的强还原剂甲脒亚磺酸(formamidine sulfinic acid, FSA).FSA分子锚固在钙钛矿薄膜的晶粒表面上阻碍锡的氧化, 导致窄带隙太阳电池的实验室PCE为21.7%(经认证PCE为20.7%), 这也是目前Sn-Pb钙钛矿单结太阳电池的最高效率.
3)大的有机阳离子.用较大的疏水性阳离子部分取代Sn-Pb钙钛矿ABX3中A位, 可以形成混合2D-3D异质结构钙钛矿, 可以有效延缓氧气和水分进入晶格[41].最有开发潜力的是仅包含几个碳原子的阳离子, 如苯乙铵(PEA)、叔丁基铵(t-BUA)等, 可以提高器件稳定性且不会对性能产生不良影响.Wei等[42]通过在反溶剂中直接引入苯乙铵配体可以实现钙钛矿膜表面和晶界处的缺陷钝化, 如图4(d)所示, 不仅改善了器件的工作稳定性, 还避免了形成过多阻碍电荷传输的层状钙钛矿.最终基于三阳离子FA/MA/Cs的Sn-Pb钙钛矿太阳电池填充因子达到79%, PCE达到19.4%(认证PCE为18.95%).Ramirez等[43]使用甲脒、铯和叔丁胺离子作为Sn-Pb钙钛矿的A位阳离子,所得钙钛矿组分为(t-BUA)2(FA0.85Cs0.15)n—1(Pb0.6Sn0.4)nI3n+1, 如图4(e)所示, 利用叔丁胺离子轴向链长短且热稳定性比其他分子高的优点, 最终基于n= 4和n= 5的Sn-Pb钙钛矿太阳电池分别实现了高达9.3%和10.6%的PCE, 并在N2中存储2000 h时分别保持了其初始PCE的47%和29%.Li等[44]向(MAPbI3)0.75(FASnI3)0.25前驱体溶液中引入少量(3.75%)的4-氟苯乙基碘化铵(FPEAI), 如图4(f)所示, 得到带隙为1.33 eV的混合2D/3D Sn-Pb钙钛矿太阳电池, 电流密度高达28.42 mA/cm2, PCE达17.51%.顶层的2D钙钛矿(1 ≤n≤ 4)能有效地抑制3D钙钛矿的相分离以及Sn2+的氧化, 从而减少了非辐射复合, 电池的开路电压提升了80 mV.

图4 (a)不同SnF2添加量的钙钛矿薄膜扫描电子显微镜(scanning electron microscope, SEM)扫描图[34]; (b) Zhu等[39]利用伽伐尼置换反应(GDR)制备MAPbxSn1—xI3钙钛矿溶液的照片和示意图以及薄膜老化过程机制示意图; (c)由于前驱体溶液中存在Sn4+而在Sn-Pb钙钛矿中形成锡空位的示意图[18]; (d) Wei等[42]利用PEAI实现Sn-Pb钙钛矿表面钝化或膜内钝化的处理方法示意图; (e) Ramirez等[43]引入叔丁胺离子n = 4和n = 5的Sn-Pb钙钛矿晶格示意图; (f) Li等[44]引入4-氟苯乙基碘化铵(FPEAI)使(MAPbI3)0.75(FASnI3)0.25晶粒高度垂直排列的示意图及未使用与使用(FPEAI)的器件J-V曲线Fig.4.(a) SEM images of perovskite films with different SnF2 additions[34]; (b) photos and schematic diagrams of preparing MAPbxSn1—xI3 precursor solution using GDR and the schematic diagram of film aging process[39]; (c) the schematic diagram of tin vacancies formation in Sn-Pb perovskite due to the presence of Sn4+ in the precursor solution[18]; (d) Wei et al.[42]used PEAI to achieve surface passivation or in-film passivation of Sn-Pb perovskit; (e) the schematic diagram of Sn-Pb perovskite lattice with n =4 and n = 5 introduced by Ramirez et al.[43]; (f) Li et al.[44]introduced FPEAI to vertically arrange the (MAPbI3)0.75(FASnI3)0.25 grain height and the J-V curve of unused and used FPEAI devices.
2.2 提高薄膜结晶质量
Sn-Pb钙钛矿薄膜形貌和质量对器件的性能具有很重要的影响.较差的表面覆盖率和多的孔洞密度会引起严重的载流子复合损失和大的漏电流.相比于铅基钙钛矿, Sn-Pb钙钛矿薄膜质量的控制难度更大.Wang等[24]通过原位表征技术探究了不同Sn-Pb比例对两步沉积的钙钛矿薄膜成膜速率及形貌的影响并结合对未退火的薄膜的SEM和XRD表征证实, 铅基前体薄膜在热退火之前形成了小尺寸的纳米晶体, 而纯锡基前体薄膜表现为更高的结晶度并在表面形成微米级的晶粒, 这表明有机组分FAI/MAI对SnI2的更大亲和力促进了室温下含锡钙钛矿的晶体生长, 从而使钙钛矿膜相比于铅基成膜迅速且晶粒尺寸增大, 如图5(a)所示.但这样的快速生长也会限制晶粒取向的自由度并抑制了在热退火过程中晶粒的重新取向, 从而导致晶粒生长方向随机, 阻碍了载流子有效传输.基于以上Sn-Pb钙钛矿薄膜结晶生长特点, 目前提出能够起到调节Sn-Pb钙钛矿薄膜结晶质量的方法主要分为四类: 1)调控组分; 2)改善薄膜制备方法; 3)溶剂工程; 4)掺杂含有功能基团的添加剂.
2.2.1 调控组分
Sn-Pb钙钛矿的晶体结构类似于铅基钙钛矿AMX3的晶体结构.M位为不同比例的Sn2+和Pb2+占据, X位通常为卤素离子Cl—, Br—或I—.A位离子位于立方晶胞的体心, M位离子位于立方晶胞的顶角, 被6个X位离子包围形成[MX6]立方八面体[45].钙钛矿结构的稳定性可以使用Goldschmidt容差因子(t)来评估, 公式如下[46]:

其中rA,rM和rX分别是A, M和X位置的离子半径.Bartel等[47]的研究表明, 稳定钙钛矿结构的最佳界线在0.825和1.059之间.通过将公差因子t调整为接近1, 晶格畸变得到有效抑制并获得更稳定的晶体结构.因此通过组分工程可以有效控制钙钛矿晶体紊乱, 提高钙钛矿薄膜结晶质量, 有利于钙钛矿材料及器件的稳定性.
目前, 在A位主要采取的离子组合有FA, MA,Cs, FA+MA, FA+Cs, FA+MA+Cs.FA+离子尺寸大于MA+, 因此FASnI3中Sn-5s和I-5p之间的反键耦合明显弱于MASnI3, 导致FASnI3中锡空位的形成能更高, 即FA+掺入可以抑制由氧气诱导的钙钛矿降解[48].但FA+的引入会对结晶度产生不利影响, 并且与MA+相比H2O和FA+之间的氢键更强, 从而加剧由水分诱导的钙钛矿降解[49].因此, 需要调控FA+及MA+离子比例以平衡由氧气和水分诱导的钙钛矿降解, 使材料及器件稳定性达到最优.为了在保持器件高性能的同时改进器件稳定性, Yang等[49]将不同量的FA+引入MAPb0.75Sn0.25I3形成MA1—yFAyPb0.75Sn0.25I3钙钛矿.最终带隙为1.33 eV的MA0.5FA0.5Pb0.75Sn0.25I3的钙钛矿太阳电池获得14.19%的稳态PCE,并在室温和相对湿度为30%—40%下储存12 d后, 仍可保留其初始PCE的80%, 在惰性气氛中储存30 d可以保留其初始PCE的94%, 如图5(b)所示.
少量Cs+取代FA+, 可提高材料热稳定性以及在空气中的稳定性, 这是由于引入少量Cs+后将引起钙钛矿立方八面体体积收缩, 从而使得FA+和I—之间的结合力增强.另外还发现Cs+的引入可以减少材料内部缺陷, 抑制载流子复合, 显著增加器件的开路电压及填充因子[50].Prasanna等[51]在A位使用FA+和Cs+, 显示比挥发性更强的MA+具有更好的热稳定性和光稳定性, 基于FA0.75Cs0.25Sn0.4Pb0.6I3的无空穴传输层(HTL-free)器件获得16.4%的PCE.且在85 ℃的黑暗环境中未封装器件1000 h以及湿热测试(85 ℃, 相对湿度为85%)1000 h下仍保持其初始PCE的95%, 在最大功率点附近和1个太阳光照射下运行1000 h以上可以保持全部初始效率.采用Cs, MA, FA组成的三阳离子结构的钙钛矿材料, 不仅可提升钙钛矿的晶相纯度, 改善其在光照及湿度环境下的稳定性, 还可以提升PCE及器件可重复性.Han等[52]基于Cs0.1MA0.2FA0.7Pb0.5Sn0.5I3的钙钛矿组分制备了以glass/ITO/NiOx/perovskite/C60/BCP/Cu为结构的器件, PCE达17.6%.
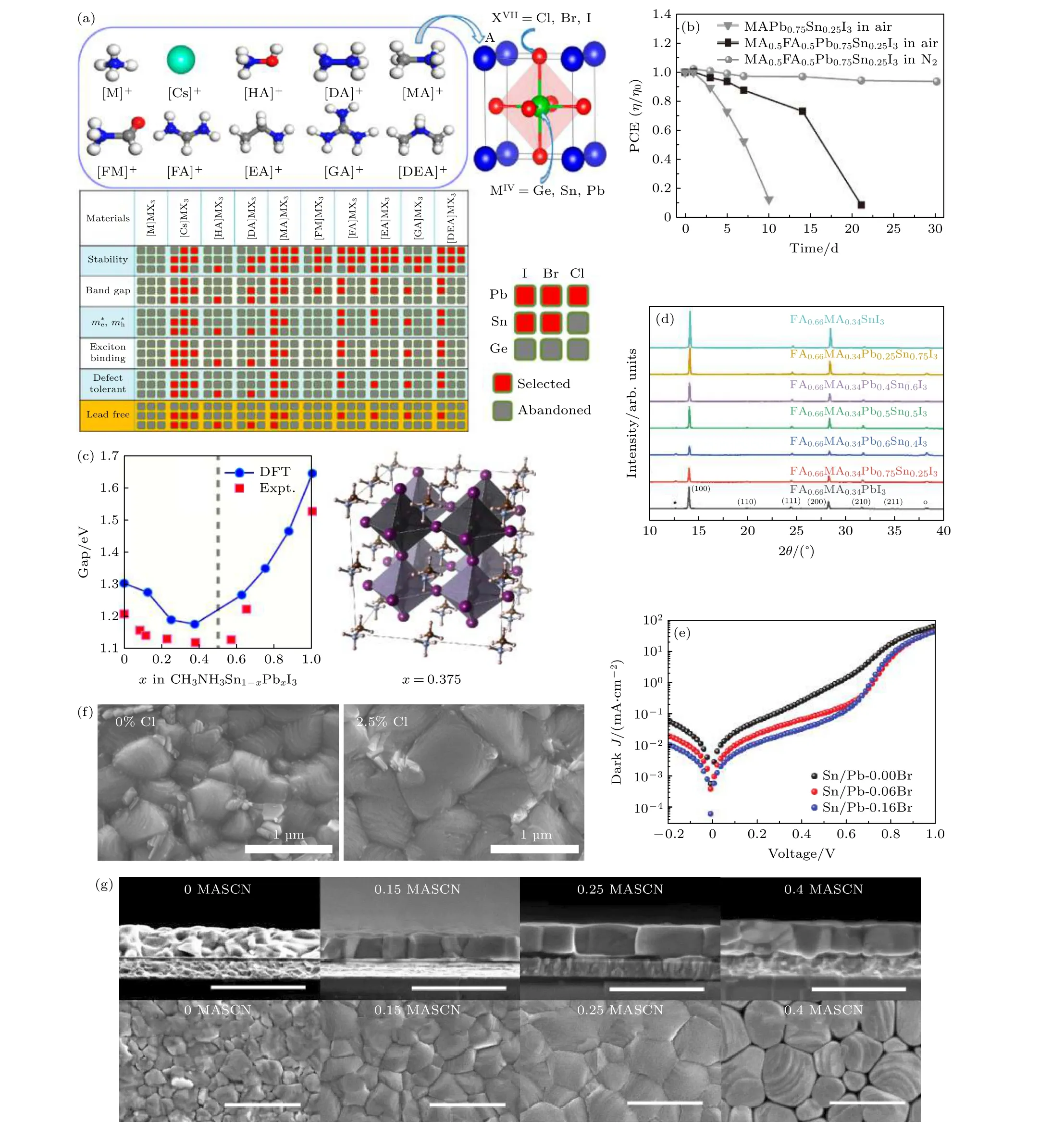
图5 (a) AMX3型钙钛矿材料常用元素组分及不同组分的材料性质[45]; (b) FA+掺入对MA1—yFAyPb0.75Sn0.25I3钙钛矿器件稳定性的影响[49]; (c)由计算和实验所得的MASn1—xPbxI3带隙随x的变化[6]; (d)不同Sn-Pb比例的FA0.66MA0.34Pb1—xSnxI3钙钛矿的XRD图谱[24]; (e) Br含量分别为0, 6%和16%的Sn-Pb钙钛矿太阳电池的暗态J-V曲线[53]; (f)未掺入Cl和掺入2.5% Cl对钙钛矿薄膜的SEM扫描图[30]; (g)掺入不同比例(0, 15%, 25%, 40%)MASCN对薄膜钙钛矿薄膜的SEM顶部扫描图及横截面扫描图[54]Fig.5.(a) The commonly used element compositions and their properties of AMX3[45]; (b) FA+-doping effects to the stability of MA1—yFAyPb0.75Sn0.25I3 perovskite devices[49]; (c) the band gap variation with x changes of MASn1—xPbxI3 obtained from calculations and experiments[6]; (d) XRD patterns of FA0.66MA0.34Pb1—xSnxI3 perovskites with different Sn-Pb ratios[24]; (e) dark J-V curves of Sn-Pb perovskite solar cells with Br concentrations of 0, 6% and 16% respectively[53]; (f) SEM images of perovskite films without Cl and with 2.5% Cl[30]; (g) the top and cross-section SEM images of perovskite films mixed with 0, 15%, 25%, 40% of MASCN[54].
对于M位的调节, 在Sn-Pb钙钛矿中显得尤为重要.锡铅比例对晶体结构、成膜速率、薄膜形貌都有显著的影响, 从而影响器件性能.随着Sn占金属比例越来越大, 带隙变窄, 也会伴随着带隙弯曲的现象.所谓带隙弯曲, 即带隙不再遵循Vegard定律, 带隙最低值一般出现在Sn含量50%—60%的情况下[7], 如图5(c)所示, 这很可能源于自旋轨道耦合(SOC)和晶格畸变之间的竞争作用[6], 但Ogomi等[5]和Hao等[8]认为是导带及价带位置变化导致的结果.最近, Klug等[9]基于FA0.83Cs0.17Pb1—ySnyI3钙钛矿将0.001%到70%的Sn替代Pb, 发现Sn含量为0.5%—20.0%范围内非辐射损失显著增加.伴随着带隙的迅速变化,当仅1%的Pb含量被Sn替代时, 光电导率、光致发光寿命和量子效率至少降低了一个数量级, 这表明低浓度的Sn掺入会导致由晶体学紊乱引起的电子缺陷, 从而引起非辐射复合损失, 器件性能严重衰减.通过优化具有30%和50% Sn组成的单结太阳电池, 分别得到了带隙为1.33 eV稳态PCE为17.6%及带隙1.30 eV稳态PCE为18.1%的窄带隙器件, 可作为底部吸收层用在全钙钛矿叠层电池中.Wang等[24]结合原位表征技术揭示了两步沉积的Sn-Pb钙钛矿薄膜结晶的时间演变过程,发现在较高的Sn含量下, 钙钛矿相的转化加速,在有机盐溶液滴加后热退火处理前可直接转变为锡基钙钛矿; 但对于仅含铅的组分, 钙钛矿相的转变会相当缓慢.因此富锡的钙钛矿薄膜结晶度要高于富铅钙钛矿, 如图5(d)所示, 这与其在室温下形成大晶粒的现象相符.
A位和M位离子也可以同时调控, Liao等[26]先分别配制FASnI3和MAPbI3的前驱体溶液, 再调控不同比例来制备(FASnI3)1—x(MAPbI3)x钙钛矿薄膜, 最终得到带隙为1.2 eV的高效(FASnI3)0.6(MAPbI3)0.4太阳电池, PCE达15.08%.
在多阳离子体系中掺杂卤素离子对X位调控,可进一步改善器件性能.目前已提出在前驱体溶液中引入卤素离子, 有效增加平均晶粒尺寸, 减少晶界密度.Li等[53]掺入溴(Br)可以有效钝化晶界, 他们发现晶界处的电荷重组是限制窄带隙Sn-Pb钙钛矿太阳电池性能的关键因素.通过优化Br浓度, 并使用一步反溶剂法结合两步退火制备出带隙为1.272 eV的(FASnI3)0.6(MAPbI3)0.34(MAPb Br3)0.06钙钛矿太阳电池, 电池暗电流密度降低2—3个数量级, 如图5(e)所示, 其开路电压损耗低至0.384 V, 填充因子高至75%, 最佳PCE大于19%.Zhao等[30]通过引入氯(Cl)来增大晶粒并减少带隙1.25 eV的Sn-Pb钙钛矿吸收层中的电子无序,如图5(f)所示, 最终得到具有18.1%的最佳PCE,在两端全钙钛矿叠层太阳电池中达到21%的PCE及20.7%的稳态PCE.
类卤素如硫氰酸盐也通常用于与SnI2前驱体形成稳定的中间络合物来延迟Sn-Pb钙钛矿的结晶.2016年, Liao等[26]使用硫氰酸铅(Pb(SCN)2)作为添加剂加入(FASnI3)0.6(MAPbI3)0.4中, 较大的晶粒可减少晶界散射, 延长载流子寿命, 并改善晶体质量.最终带隙约1.2 eV的窄带隙Sn-Pb钙钛矿太阳电池的最佳认证PCE为17.01%.之后Lian等[54]还使用硫氰酸甲铵(MASCN)作为双功能添加剂沉积了高质量的FAPb0.7Sn0.3I3钙钛矿薄膜.MASCN可以调钙钛矿薄膜的形态, 如图5(g)所示, 而且通过SCN—和Sn2+之间的强配位作用延迟前驱体溶液中Sn2+的氧化.最终基于FAPb0.7Sn0.3I3的Sn-Pb钙钛矿太阳电池在添加25% MASCN的情况下获得79%的高填充因子和16.26%的最佳PCE.2019年, Tong等[55]向约1.25 eV带隙的(FASnI3)0.6(MAPbI3)0.4钙钛矿中添加硫氰酸胍(GuaSCN), 发现其可以在晶界处形成二维(2D)结构, 通过其更宽的带隙钝化晶界和表面, 改善钙钛矿的电子性能; 并且能够阻止晶粒中的锡扩散以及抑制过量的锡空位的形成; 同时还可以减少氧通过表面和晶界扩散到晶粒中, 以抑制Sn2+的进一步氧化.最终使薄膜中缺陷密度降低, 载流子寿命超过1 μm, 扩散长度为2.5 μm, 从而获得20.5%的最佳PCE; 与宽带隙钙钛矿太阳电池结合使用时, 获得PCE为25%的高效四端和23.1%的两端全钙钛矿叠层电池.
2.2.2 改善薄膜制备方法
Sn-Pb钙钛矿薄膜制备方法的改善也可有效提高薄膜结晶质量.传统的一步反溶剂法通常以N, N-二甲基甲酰胺(DMF)和二甲基亚砜(DMSO)作为溶剂, 在溶液旋涂时滴加氯苯(Ph-Cl)、甲苯(Ph-Me)或二乙醚(DE)之类的反溶剂.反溶剂会迅速从湿膜中除去前驱体溶剂并引发多晶钙钛矿薄膜的快速成核和晶体生长, 正因为如此, 使用一步反溶剂法很容易产生仅几百纳米的小晶粒钙钛矿[22,56].这些小晶粒伴随着大量晶界的形成, 而晶界被认为是电荷载流子非辐射复合中心, 所以晶界密度的增加会使非辐射复合损耗严重, 显著降低器件开路电压, 影响器件光伏性能[53].最近, Wei等[42]提出在反溶剂中直接引入苯乙铵(PEAI)配体, 这种大的有机阳离子可以在表面形成超薄的2D钝化层, 可实现钙钛矿膜表面和晶界的缺陷钝化.这种膜内钝化方法还改善了器件的工作稳定性, 同时还避免了过多层状钙钛矿的形成以阻碍电荷的传输.最终基于Cs0.1MA0.2FA0.7Pb0.5Sn0.5I3组分钙钛矿的器件填充因子为79%, 认证PCE高达18.95%.
两步沉积法也可用于制备Sn-Pb钙钛矿薄膜.通常需先使用DMF或DMSO溶剂溶解SnI2和PbI2, 然后将前驱体溶液旋涂在具有空穴传输层的衬底上.再将含有碘甲胺(CH3NH3I, MAI)或其他有机盐的异丙醇(IPA)溶液滴在中间体薄膜上使有机盐和无机膜之间反应得到钙钛矿相.有研究表明, 由于SnI2在IPA中的高溶解度[57,58]并且SnI2与有机盐的快速反应[25], 导致两步顺序沉积很少应用在Sn-Pb钙钛矿薄膜的制备中.但这并不表示两步顺序沉积法不可应用于制备Sn-Pb钙钛矿薄膜.早在2016年, Zhu等[59]使用两步顺序沉积结合DMSO溶剂蒸气处理的方式, 得到晶粒尺寸大且光滑的高质量薄膜, 如图6(a)所示, 最终基于MASn0.1Pb0.9I3的Sn-Pb钙钛矿太阳电池获得了最高10.25%的PCE.2019年, Lian等[54]通过两步顺序沉积制备FA基Sn-Pb钙钛矿薄膜, 获得16.26%的PCE, 也是目前基于FA基的Sn-Pb钙钛矿太阳电池的最高PCE.最近, Wang等[24]使用顺序沉积法制备窄带隙1.23 eV的FA0.66MA0.34Pb0.5Sn0.5I3钙钛矿太阳电池, 并结合原位吸收光谱表征手段, 如图6(b)所示, 最终获得稳态PCE为16.1%的钙钛矿太阳电池.能够通过两步顺序沉积方法得到这些高质量结晶的锡铅钙钛矿薄膜, 主要是由于DMSO与前驱体溶液中的SnI2和PbI2有较强的配位作用, 导致中间体薄膜以非晶态的形式存在[23], 这不仅有利于后续有机盐与之反应, 还可以有效调控成膜动力学, 从而制备平滑且致密的钙钛矿薄膜.

图6 (a)两步顺序沉积结合DMSO溶剂蒸气处理的方式制备MASn0.1Pb0.9I3的过程示意图[59]; (b)两步顺序沉积FA0.66MA0.34 Pb0.5Sn0.5I3的过程示意及原位吸收光谱图[24]Fig.6.(a) The schematic diagram of the two-step sequential depositions combined with DMSO solvent vapor treatment method to prepare MASn0.1Pb0.9I3 perovskite films[59]; (b) the schematic diagram of the two-step sequential deposition process of FA0.66MA0.34Pb0.5Sn0.5I3 and in-situ absorption spectra[24].
除传统的溶液法, 真空辅助法和蒸发法也可以用于Sn-Pb钙钛矿薄膜的制备.真空辅助方法可以去除新鲜沉积的钙钛矿湿膜中多余的溶剂来得到中间相, 从而对成膜结晶动力学起到有效调控作用.目前真空辅助可结合溶液旋涂法、刮涂法等.2018年, Liu等[60]将真空辅助热退火结合一步溶液法, 为制备高覆盖率和高结晶度的MASn0.5Pb0.5IxCl3—x窄带隙钙钛矿薄膜提供了一种简单的方法,所得器件PCE超过12%.通过观察在真空和氮气环境下退火所得的薄膜形貌, 可以表明该方法还可以加速甲基氯化铵(MACl)的升华从而降低钙钛矿薄膜中的陷阱密度, 如图7(a)和(b)所示.Nejand等[61]研究发现真空辅助生长(vacuum-assisted growth, VAGC)方法可显著延长电荷载流子寿命,薄膜呈现出大的柱状晶粒, 几乎没有垂直于薄膜表面的晶界, 如图7(c)所示, 有利于载流子的有效传输.基于FA0.8MA0.2Sn0.5Pb0.5I3的钙钛矿太阳电池的PCE达到18.2%, 作为窄带隙底电池应用于四端全钙钛矿叠层太阳电池PCE高达23%.最近,Li等[44]首次报道了基于真空辅助刮涂法制备高质量Sn-Pb钙钛矿薄膜, 获得混合2D/3D Sn-Pb钙钛矿太阳电池PCE可达17.50%.相比于传统的高温刮涂法, 该方法增加真空预结晶过程, 使前体膜沉积过程与随后的退火过程分离, 延缓原本快速的晶体成核与生长过程, 为未来Sn-Pb钙钛矿太阳电池大面积的制备提供思路.
蒸发法可以有效避免在钙钛矿层沉积过程中对其他器件层的损坏, 且相比于传统溶液法更容易得到均匀致密的高质量薄膜.但目前应用于制备Sn-Pb钙钛矿薄膜的研究较少.2019年, Ball等[62]提出了一种双源共蒸发法制备窄带隙FA1—xCsxSn1—yPbyI3钙钛矿薄膜的方法.他们使用由Cs, Pb和Sn金属卤化物熔融形成的混合物作为阳离子的单坩埚源, 然后与碘甲脒(FAI)共蒸发, 如图7(d)所示, 获得PCE为10%的器件性能.
2.2.3 溶剂工程

图7 (a)真空辅助热退火结合一步溶液法制备的器件结构及原理示意图[60]; (b)使用/不使用真空辅助热退火所制备的薄膜形貌顶部SEM图[60]; (c)真空辅助生长(VAGC)方法的原理示意图及横截面SEM图像[61]; (d)双源共蒸法制备FA1—xCsxSn1—y PbyI3钙钛矿薄膜过程示意图及晶体结构示意图[62]Fig.7.(a) Device architecture and schematic diagram that combined the vacuum-assisted thermal annealing process and one-step solution method[60]; (b) the top SEM images of the film prepared with/without vacuum-assisted thermal annealing[60]; (c) the schematic diagram and cross-section SEM image of the film prepared by VAGC method[61]; (d) the schematic diagram and crystal structure of FA1—xCsxSn1—yPbyI3 perovskite films prepared by dual-source co-evaporation method[62].
正如前述, DMSO与前驱体溶液中的SnI2和PbI2有较强的配位作用, 因此在2017年, Zhu等[56]简单地通过调节前驱体溶液中的DMSO的量来实现控制MASn0.25Pb0.75I3钙钛矿薄膜的结晶.他们发现减少混合溶剂中DMSO的量会使钙钛矿相形成的更快, 这是由于DMF的键合力较弱且挥发性更大, 因此一定量的DMSO可利于MASn0.25Pb0.75I3钙钛矿薄膜表现出显著的(110)择优取向, 质构系数提高了2.6倍.同时发现DMSO的引入避免了在长期储存的前驱体中观察到的胶体凝结的形成,如图8(a)所示, 并改善了钙钛矿薄膜中锡/铅的不均匀分布.但过量的DMSO将PbI2和SnI2胶簇溶解成较小的尺寸, 从而降低钙钛矿的结晶度[63].最终通过优化溶剂比例及器件工艺, 实现无迟滞的MASn0.25Pb0.75I3钙钛矿获得15.2%的PCE.Liu等[64]发现PbI2/SnI2可以通过DMSO溶剂插入,从而形成PbI2/(SnI2) (DMSO)x络合物.在络合过程中DMSO溶剂有利于无定形镜状膜的形成, 同时当PbI2/SnI2与DMSO络合时, 由于SnI2其较高的活化能, 因此可控制络合过程.之后由于MAI与PbI2/SnI2的更高亲和能, 将与DMSO交换从而形成具有高密度和完全覆盖率的MAPb1—xSnxI3薄膜, 如图8(b)所示.通过不同的x值可调节带隙从而可以实现高效的Sn-Pb钙钛矿太阳电池, 最大PCE为14.12%.之后他们基于此项工作, 又向MAPb0.75Sn0.25I3钙钛矿前驱体溶液中掺杂少量的C60来制备Sn-Pb钙钛矿薄膜[65].C60和DMSO分子之间存在的结合能会调节钙钛矿的结晶过程,这会导致钙钛矿薄膜针孔减少并且C60在晶界处均匀分布, 如图8(c)所示, 增加了本体和表面载流子复合寿命, 并降低了电荷陷阱态密度, 电池显示出更高的PCE、更小的迟滞效应和更好的长期稳定性.因此, 不同组分的锡铅钙钛矿材料在制备过程中所选用的溶剂也不同, 需要根据有机盐及金属碘化物的具体情况以调控DMF与DMSO的比例,使DMSO发挥适度的键合能力, 有效调控锡铅钙钛矿薄膜的生长; 另外, 还可以在前驱体溶液中添加富勒烯类化合物, 不仅调节结晶, 还能够对晶界起到钝化作用.
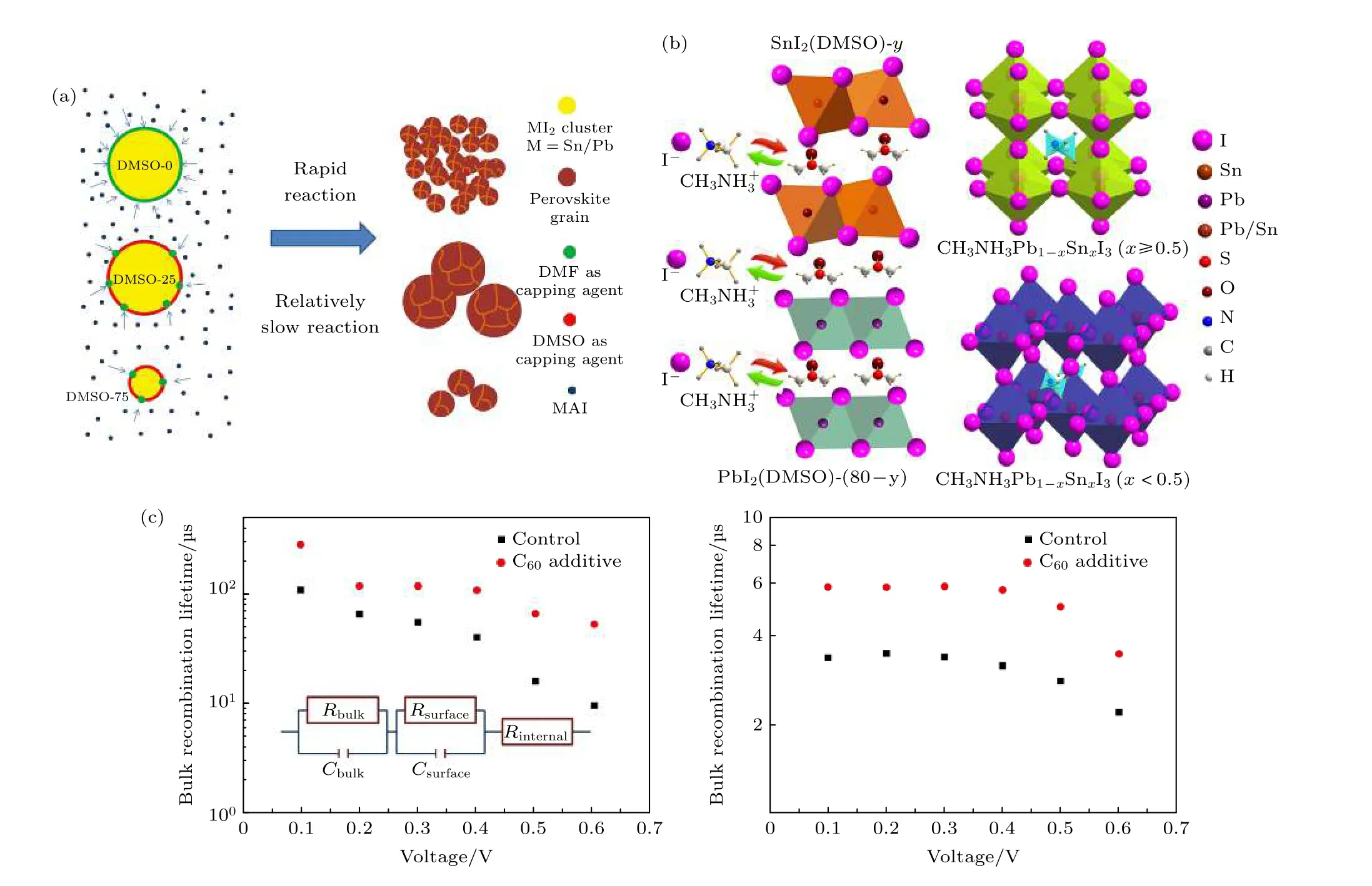
图8 (a)不同DMSO/DMF溶剂比的Sn-Pb钙钛矿反应机理示意图[56]; (b) MAPb1—xSnxI3 (0 ≤ x ≤ 1)薄膜的形成机理以及相应的晶体结构示意图[64]; (c)在不同偏置电压下未掺杂及掺杂C60的MAPb0.75Sn0.25I3钙钛矿器件的本体复合寿命和表面复合寿命[65]Fig.8.(a) The mechanism diagram of Sn-Pb perovskite reactions with different DMSO/DMF solvent ratio[56]; (b) the formation mechanisms of MAPb1—xSnxI3 (0 ≤ x ≤ 1) film and corresponding crystal structures[64]; (c) bulk recombination life and surface recombination life of MAPb0.75Sn0.25I3 perovskite devices with or without C60-doped under different amplitude voltages[65].
2.2.4 掺杂含有功能基团的添加剂
还有一些添加剂, 由于含有一些特殊的功能基团, 如含有羟基(—OH)、氨基(—NH2)等可形成氢键的化合物或(及)含有羰基(—C=O)、氰基(—C≡N)、卤素离子等具有非键合电子的路易斯碱, 也可以对Sn-Pb钙钛矿的结晶过程进行调控,从而有效改善钙钛矿结晶质量.Zhou等[66]将溴化胍(GABr)引入钙钛矿膜中, 如图9(a)所示, 用胍离子()部分取代FA+和MA+, 更多氨基所产生的更强的氢键相互作用可钝化晶界并抑制空位缺陷的形成, 另外中三个均匀分布的氨基使得其具有零偶极矩的特性, 这被证明有利于消除Sn-Pb钙钛矿电池中的迟滞现象.同时Br—的引入通常会增加带隙, 还可以改善结晶度和增大晶粒尺寸、减少无序性和钝化缺陷, 促进电荷提取和提高环境稳定性.最终在1.35 eV带隙的FA0.7MA0.3Pb0.7Sn0.3I3钙钛矿太阳电池中实现了20.63%的最佳PCE和19.8%的认证PCE, 开路电压损耗仅为0.33 V.最终在钙钛矿薄膜优化厚度为1000 nm的情况下, 单结窄带隙钙钛矿太阳电池和钙钛矿/钙钛矿叠层电池分别实现了20.2%和22.7%的稳态PCE, 如图9(b)所示.最近, Hu等[67]提出在纯无机Sn-Pb钙钛矿CsPb0.6Sn0.4I3薄膜的上表面旋涂有机盐(氨基甲基)哌啶二碘化物(4AMP)I2.大的有机阳离子(4AMP)2+不仅钝化表面缺陷, 而且形成疏水性包封层以防止钙钛矿薄膜受潮.同时他们发现—和官能团的组合可能是关键,因为用哌嗪(带有两个=基团)碘化物或苯乙铵(仅含—)碘化物代替(4AMP)I2均会导致较低的PCE, 如图9(c)所示.最终实现带隙为1.38 eV的CsPb0.6Sn0.4I3钙钛矿的器件PCE高达13.37%,并且表现出很好的工作稳定性.另外, 引入不同尺寸的离子进入晶格中也可以有效调节钙钛矿晶格结构, 减少由晶格紊乱所产生的缺陷位点, 提高钙钛矿结晶质量.Yang等[68]将CdI2添加到FA0.5MA0.45Cs0.05Pb0.5Sn0.5I3钙钛矿前驱体溶液中, 使离子半径相对较小的Cd2+掺入钙钛矿的晶格中.他们发现Sn-Pb钙钛矿太阳电池中的电荷收集效率受到电子扩散长度的限制, 而这一策略可有效填补Sn空位, 从而提高少子的复合寿命和载流子迁移率, 电子扩散长度长达2.72 ± 0.15 μm.
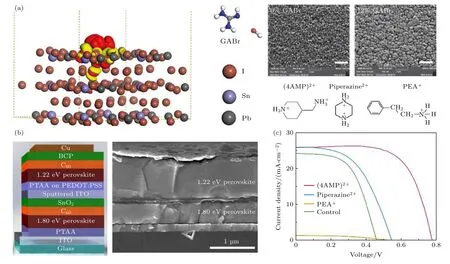
图9 (a)含GABr的FA0.7MA0.3Pb0.7Sn0.3I3表面的电荷密度分布图(等电势为0.03 eÅ—3)以及未添加与添加12%的GABr的钙钛矿SEM扫描图[66]; (b)添加CdI2的1.22 eV窄带隙钙钛矿与1.80 eV宽带隙钙钛矿叠层太阳电池的结构示意图和SEM横截面扫描图[68]; (c) (4AMP)2+, 哌嗪离子和PEA+阳离子的结构式及用(4AMP)I2, 碘化哌嗪和PEAI表面处理的CsPb0.6Sn0.4I3钙钛矿太阳电池的J-V曲线[67]Fig.9.(a) The charge density distribution on the FA0.7MA0.3Pb0.7Sn0.3I3 surface containing GABr (the isopotential is 0.03 eÅ—3) and the SEM images of the perovskite without and with 12% GABr[66]; (b) structure diagram and cross-section SEM inage of 1.22 eV narrow-bandgap perovskite with CdI2 added and 1.80 eV wide-bandgap perovskite tandem solar cell[68]; (c) structure of(4AMP)2+,piperazine ion and PEA+ and J-V curves of CsPb0.6Sn0.4I3 perovskite solar cell that absorber film surface treated with (4AMP)I2,piperazine iodide and PEAI, respectively[67].
2.3 选用合适的载流子传输层
Sn-Pb钙钛矿太阳电池的开路电压较小, 主要原因是由于其开路电压损耗严重, 该损耗主要分为三部分: 第一部分为钙钛矿吸收层内部, 第二部分为钙钛矿和电子传输层界面处, 第三部分在钙钛矿和空穴传输层界面处.因此, 选用合适的载流子传输层, 提高界面能级匹配程度, 可有效减少Sn-Pb钙钛矿太阳电池界面处的开路电压损耗, 提高器件性能.
电子传输层(electron transport layer, ETL).Kapil等[69]借鉴了铜铟镓硒(CIGS)太阳电池中缓冲层和吸收层之间的导带偏移理论[70], 认为0—0.3 eV的导带偏移量有助于减少界面处的载流子复合以获得更高的开路电压.因此他们在FA0.5MA0.5Sn0.5Pb0.5I3钙钛矿吸收层的顶部引入一层约25 nm薄的苯基—C60—丁酸甲酯(PCBM),导带偏移0.15 eV, 如图10(a)所示, 这种尖峰结构可以改善界面电荷的流动, 使界面陷阱以及载流子复 合 减 少, 从 而 提 高 开 路 电 压 至0.75 V, 最 佳PCE为17.6%.但是考虑到器件制备的成本问题,Zhou等[71]开发了新型的S-乙酰硫代胆碱氯化物分子取代了昂贵的PCBM, 并且能有效钝化钙钛矿中的正负电荷离子缺陷.并且与PCBM相比,经S-乙酰硫代胆碱氯化物分子钝化的器件还具有更好的环境稳定性.除了研发新的电子传输材料及结构, 还可以利用界面工程对Sn-Pb钙钛矿层及电子传输层之间的界面处理, 减少界面陷阱态密度, 改善器件光伏性能.Liu等[72]开发了氟烷基取代的富勒烯DF-C60, 利用氟烷基官能团的低表面能, 在MAPb0.5Sn0.5I3中形成梯度异质结(gradient heterojunction, GHJ)结构, 如图10(b)所示, 可以起到钝化缺陷的作用, 并能够降低膜中Urbach能量, 减少陷阱密度、双分子复合以及表面/本体复合.此外, DF-C60还可以增加Sn-Pb钙钛矿表面疏水性, 提高器件整体稳定性.最终获得高达0.89 V的开路电压及15.61%的PCE.
空穴传输层(hole transport layer, HTL).PEDOT:PSS是在目前高效的窄带隙钙钛矿太阳电池中广泛使用的空穴传输层[18,73,74].但Prasanna等[51]的研究工作表明, PEDOT:PSS与钙钛矿之间存在相互反应, 导致热老化后电荷提取严重恶化, 限制了钙钛矿太阳电池的稳定性.因此他们使用ITO-钙钛矿异质结(无空穴传输层)结构制备了完整的FA0.75Cs0.25Sn0.4Pb0.6I3钙钛矿太阳电池, 实现热稳定性并在85 ℃的条件下老化1000 h维持初始性能的95%.Yang等[75]基于带隙1.34 eV的纯无机CsSn0.3Pb0.7I3钙钛矿吸收层, 也分别制备了以PEDOT:PSS为空穴传输层和无空穴传输层的完整器件, 分别达到9.41%和7.60%的PCE, 是目前带隙低于1.40 eV的无机PSCs的最高PCE.
除了采用无空穴传输层的结构, 开发无机空穴传输材料也是替代PEDOT:PSS的有效方法.2018年, Chi等[27]发现PEDOT:PSS可能不是Sn-Pb钙钛矿太阳电池的理想空穴传输层主要归因于PEDOT:PSS的高度亲水性导致的钙钛矿膜形貌差和/或界面能级对准较差[76—79].对此他们使用300 ℃高温处理的NiOx代替PEDOT:PSS, 减少表面润湿性以限制膜上成核位点的分布, 从而改善钙钛矿结晶质量并增强电荷提取, 如图10(c)所示.更重要的是, 所制备的器件有非常好的热稳定性.最终基于FAPb0.75Sn0.25I3的纯FA基Sn-Pb钙钛矿太阳电池获得17.25%的PCE.2019年, Han等[52]首次报道了低温溶液处理的NiOx膜作为Sn-Pb钙钛矿太阳电池的空穴传输层, 具有3.4 nm的低表面粗糙度及可忽略的寄生吸收, 并且该膜的功函数为4.72 eV, 价带能级为5.17 eV, 与Cs0.1MA0.2FA0.7Pb0.5Sn0.5I3钙钛矿有良好的能级对准性.最终实现31 mA cm—2的短路电流密度及17.6%的高PCE(稳态PCE为17.0%), 并且在存储102天后仍保留其初始性能的95%.最近, Yu等[80]利用低温原子层沉积(LT-ALD)方法得到不完全氧化的SnO1.76层, 具有双极性载流子传输特性, 能够得到与使用PEDOT:PSS几乎相同的光电流且具有更好的器件稳定性, PCE达20.2%.

图10 (a) Kapil等[69]对比传统无PCBM层和带PCBM层的电荷提取和复合过程示意图, τr表示从FAMA到C60的载流子注入时间; (b)添加DF-C60形成的梯度异质结(GHJ)结构示意图[72]; (c)在NiOx及PEDOT:PSS上沉积钙钛矿膜的SEM顶部扫描图及截面扫描图[27]; (d)使用BHJ PBDB-T:ITIC中间层形成的逐步升高的HOMO能级结构示意图[82]; (e) S-乙酰硫代胆碱氯化物分子锚定在缺陷部位的示意图, 其中红色、黄色和蓝色符号分别代表S-乙酰硫代胆碱氯化物分子中的O原子、S原子和N原子[71]Fig.10.(a) The diagram of Kapil et al[69].compared the traditional charge extraction and recombination process without and with PCBM, τr represents the carrier injection time from FAMA to C60; (b) the schematic diagram of the gradient heterojunction (GHJ)with DF-C60[72]; (c) the top and cross-section SEM images of the perovskite films deposited on NiOx and PEDOT:PSS[27]; (d) the schematic diagram of the gradually increasing HOMO energy level structure formed by BHJ PBDB-T:ITIC intermediate layer[82];(e) the schematic diagram of the S-acetylthiocholine chloride molecule anchored at the defect sites, where the red, yellow and blue symbols represent the O atom, S atom and N atom in the acetylthiocholine chloride molecule, respectively[71].
对PEDOT:PSS的掺杂改性也可以有效改善空穴传输层与钙钛矿界面的能级匹配情况以及钝化界面陷阱态.2018年, Tang等[81]通过用全氟化离聚物PFI掺杂PEDOT:PSS, 功函数从—5.02 eV调整至—5.19 eV, 这样可以减少FA0.6MA0.4Sn0.6Pb0.4I3吸收层和空穴传输层之间的能级失配, 从而将开路电压提高50 mV以上, 最高达0.783 V, PCE提高至15.85%且显示出更高的稳定性.Xu等[82]使用超薄异质结(bulk heterojunction, BHJ)有机半导体PBDB-T:ITIC作为PEDOT:PSS与(FASnI3)0.6(MAPbI3)0.4膜的中间层.该BHJ PBDBT:ITIC中间层具有逐步升高的HOMO能级和有效的载流子抽取/传输能力, 如图10(d)所示, 因此它可以形成梯度能带对准和有效的电荷传输通道.重要的是, 这种BHJ PBDB-T:ITIC层也可以促进Sn-Pb钙钛矿的高质量生长, 并且其CN和O原子还可以有效地钝化膜表面的陷阱态密度.最终钙钛矿器件PCE为18.03%, 开路电压损失仅有0.39 V, 且器件稳定性和可重复性也表现出显著的提高.2019年, Zhou等[71]使用EMIC(氯化1-乙基-3-甲基咪唑)离子液体对PEDOT:PSS进行改性, 如图10(e)所示, 从而获得具有高电导率、低功函和光滑表面的空穴传输层并应用于铅基钙钛矿太阳电池中.之后他们将此空穴传输层应用于带隙为1.34 eV的FA0.7MA0.3Pb0.7Sn0.3I3钙钛矿太阳电池中, 在未添加其他添加剂的情况下, 能实现0.91 V的高开路电压[66].
3 总结及展望
目前Sn-Pb钙钛矿太阳电池的短路电流密度一直是很高的水平, 限制其性能提高的主要因素是较高的开路电压损耗及器件的不稳定性.本文基于以上问题, 主要讨论了Sn2+易氧化为Sn4+所引起的重p掺杂; SnI2与有机碘化物的快速反应导致的难以控制的薄膜结晶过程以及载流子传输层不合适所造成的界面能级不匹配或器件的不稳定性这三方面问题.针对以上问题, 本文综述了近年来提出的有效解决策略.
对于Sn-Pb钙钛矿中Sn2+氧化问题, 首先明确了Sn-Pb钙钛矿的氧化机制并提出抗氧化的几种方法, 如调控Sn-Pb比例和添加富锡化合物降低Sn2+氧化趋势, 使用含有烷羟基(—R—OH)、酚羟基(—Ph—OH)、次磷酸根()等还原性基团的化合物或能与Sn(Ⅳ)发生归中反应的Sn(0)物质将氧化的Sn2+还原, 以及添加大的有机阳离子形成混合2D-3D异质结构有效阻挡氧气与水分的进入.对于Sn-Pb钙钛矿中薄膜形貌和结晶质量问题, 对比了Sn-Pb钙钛矿薄膜与铅基钙钛矿的结晶生长过程, 明确了Sn-Pb钙钛矿薄膜更快速的结晶过程是由于Sn2+与有机碘化物更强的亲和力引起的, 同时Sn2+与Pb2+的混合B位也导致Sn-Pb钙钛矿晶格的缺陷容忍度明显低于铅基钙钛矿.对此本文总结了几种能够起到调节Sn-Pb钙钛矿薄膜结晶质量的方法, 如调控组分以有效控制钙钛矿晶体紊乱, 改变薄膜制备方法、通过溶剂工程以及添加含有羟基(—OH)、氨基(—NH2)等可形成氢键的化合物或(及)含有羰基(—C=O)、氰基(—C≡N)、卤素离子等具有非键合电子的路易斯碱的功能性添加剂有效延缓结晶生长速度; 对于Sn-Pb钙钛矿中载流子传输层所引起的界面能级失配或器件不稳定的问题, 本文也给出了几种可替代的有效的载流子传输层, 如PCBM-C60双电子传输层或无机空穴传输层NiOx等, 界面能级对准情况的改善能保证载流子更有效的抽取分离; 或对传统的载流子传输层进行掺杂优化, 如向电子传输层C60中添加富勒烯类衍生物, 既能改善界面能级失配问题, 又能有效钝化钙钛矿晶界缺陷, 有效减少Sn-Pb钙钛矿太阳电池界面处的开路电压损耗, 提高器件性能.
对于未来Sn-Pb钙钛矿太阳电池的发展, 有以下几点展望.
1)铅基钙钛矿的缺陷态密度范围为1013—1014cm—3[83,84], 但Sn-Pb钙钛矿的缺陷态密度要高2—3个数量级.目前国内外研究人员对Sn-Pb钙钛矿中的缺陷形成机理认识的还不够全面深入,未来还需要结合纯铅基或纯锡基钙钛矿太阳电池的研究, 进一步制定添加剂策略和相应的缺陷钝化机制.需要更好地理解缺陷钝化机制和明确关键的功能基团, 指导设计添加剂分子以及多功能添加剂以发挥协同钝化作用.
2)为了制备大尺寸、高结晶质量的Sn-Pb钙钛矿, 要更加深入理解其结晶动力学, 增加晶粒的纵向生长, 从而促进载流子的有效传输.其快速结晶所导致的孔洞、裂纹等3D缺陷会严重降低载流子扩散长度、增加载流子复合, 影响器件性能.开发新的添加剂或新的薄膜制备方法以及利用溶剂工程等控制钙钛矿结晶动力学将是有效的方法; 在保证结晶质量的前提下可再适度增加薄膜厚度,使Sn-Pb钙钛矿太阳电池的短路电流密度进一步提高.
3) Sn-Pb钙钛矿中Sn2+氧化引起的器件不稳定性问题是一个巨大的挑战.为避免发生在SnI2固体及锡铅钙钛矿前体溶液中的氧化, 锡源净化是有必要的, 要保证源材料中Sn2+的纯度或寻求有效的还原剂添加至Sn-Pb钙钛矿前体溶液中; 为避免钙钛矿沉积结晶过程中氧分子的吸附可借助惰性气氛或开发新型溶剂; 并且, 有效的封装也可以抵抗氧气和水分的侵入, 防止Sn-Pb钙钛矿的分解.
4)对载流子传输层的研究在Sn-Pb钙钛矿太阳电池中也是非常重要的.一方面其能带结构区别于铅基钙钛矿, 传统载流子传输层与之可能存在一定的能级势垒, 影响载流子有效的传输, 因此要明确不同组分Sn-Pb钙钛矿的能带位置, 寻求能够使能级移动的修饰材料或能级更为匹配的新型载流子传输材料; 另一方面, 载流子传输层本身的性质(如酸性、吸湿性等)也会导致器件的不稳定性,在界面使用钝化材料对其改性可能会是行之有效的方法.
总之, Sn-Pb钙钛矿太阳电池的几个主要问题在未来解决之后, 器件效率有望与铅基钙钛矿太阳电池并肩齐驱, 实现高效稳定的单结太阳电池, 同时还可以应用于高效全钙钛矿叠层太阳电池, 助力实现钙钛矿太阳电池的商业可行性.
附录
猜你喜欢
物理学报(2023年3期)2023-02-19 08:09:20
物理学报(2022年6期)2022-03-30 14:27:14
数字技术与应用(2021年2期)2021-04-22 03:22:24
陶瓷学报(2020年5期)2020-11-09 09:22:54
通信电源技术(2018年5期)2018-08-23 01:16:40
电子技术与软件工程(2017年19期)2017-11-09 11:05:47
材料科学与工程学报(2016年5期)2016-02-27 07:11:17
云南师范大学学报(自然科学版)(2015年5期)2015-12-26 12:46:14
电源技术(2015年5期)2015-08-22 11:18:12
电源技术(2015年7期)2015-08-22 08:49:00
